
达沙替尼衍生物的合成及其产率分析
2021-09-09 01:39王延玲袁琦蒲晓辉
河南大学学报(医学版) 2021年4期
王延玲袁 琦蒲晓辉
河南大学 药物研究所,河南 开封475004
基于在临床试验中获得的有效数据,达沙替尼(DAS)于2006 年被FDA 批准用于治疗BCR⁃ABL依赖的慢性髓系白血病或费城染色体阳性的对伊马替尼治疗有抗药性或不耐受的成人急性淋巴细胞白血病[1⁃3]。
肿瘤治疗的一个新兴领域是针对肿瘤微环境,而DAS 是一种强效的BCR⁃ABL 激酶抑制剂,在0.6~0.8 mmol 的浓度下仍然可以发挥作用。并且DAS可干扰与血管生成和人类癌细胞转移级联有关的关键细胞功能,如运动和侵袭。DAS 通过直接影响血管内皮细胞和抑制某些肿瘤细胞的促血管生成信号,降低了微血管内皮细胞在体外形成血管的能力,以及肿瘤细胞诱导周围的血管生成能力,从而抑制肿瘤生长[4⁃6]。
由于DAS 独特的抗癌机制和低毒性,已成为治疗慢性粒细胞白血病(chronic myelogenous leukemia,CML)的最佳药物。除此之外,DAS 在治疗各种癌症中,如白血病、肺癌、前列腺癌和卵巢癌,也取得了令人鼓舞的结果[7⁃9]。然而,DAS 的极低溶解性导致其应用受限,且有试验表明,DAS 虽未发现耐药性,但在使用过程中依然存在胸膜积液、发热、血小板减少、心脏衰竭,呼吸困难等不良反应[10⁃12]。因此,对DAS 进行适当的修饰,可以改善其溶解性,最大限度地降低全身毒性,从而提高治疗效率。因此,本研究利用DAS 上的羟基与丁二酸酐(SA)开环后的羧基进行酯化反应,制备带有游离羧基的达沙替尼丁二酸单酯衍生物(DAS-SA),并采用高效液相色谱法(HPLC)来间接测定产物中的DAS⁃SA。根据标准曲线法计算产物中DAS⁃SA 的百分含量,并对该反应的产率进行计算,同时为后期深入研究DAS 的衍生物的含量测定提供方法依据,也为进一步修饰DAS 以改善其理化性质和成药性奠定基础。
1 仪器和试剂
1.1 仪器
磁力搅拌器(上海力辰科技有限公司),紫外可见分光光度计(UV⁃2600,日本岛津公司),Waters2695⁃2489 型高效液相色谱仪(沃特世科技上海有限公司),分析天平(BSA224S,德国Sartorius)。
1.2 试剂
达沙替尼原料药(DAS,批号181015,南京德尔诺医药科技有限公司);达沙替尼对照品(批号420041⁃201601,纯度97%,中国食品药品检定研究院);丁二酸酐(SA,批号20180129,国药集团化学试剂有限公司);甲醇(批号600722⁃07813,色谱纯,天津市四有精细化学品有限公司);乙腈(批号20180131,色谱纯,天津市科密欧化学试剂有限公司);二甲基亚砜(DMSO,批号20191005,分析纯,天津市德恩化学试剂有限公司);N,N⁃二甲基甲酰胺(DMF,批号20180108,徐州天鸿化工有限公司)。水为哇哈哈纯净水,其他试剂均为分析纯。
2 方法与结果
2.1 达沙替尼丁二酸单酯衍生物的合成
DAS 0.1 mmol,SA 0.5 mmol,置于三颈瓶中加入3 mL DMF 在N2气中搅拌溶解。然后加入1 mL 三乙胺,在N2气中继续反应48 h(25 ℃,500 r/min)。反应结束后,将反应液旋至糖稀状,用CH2Cl2和CH3OH 洗涤,离心得到沉淀,真空干燥即得DAS⁃SA。
2.2 衍生物的核磁共振氢谱(1H NMR)表征
将适量(10~20 mg)的待测样品溶解于氘代二甲基亚砜(DMSO⁃d6)中,以四甲基硅烷(TMS)作内标,在室温下,用核磁共振仪测定其核磁共振氢谱(1H NMR)。
SA、DAS 及DAS⁃SA 的1H NMR 图所示,在SA 图谱中δ=3.01×10-6处为SA 中亚甲基(-CH2)的化学位移。在DAS 图谱中,δ=7.17、7.53×10-6处为DAS 苯环上的质子峰,δ=8.47、10.21×10-6,为DAS 上b、c(-NH-)的质子峰,a处为DAS 上羟基中的质子的化学位移。在DAS⁃SA 的图谱中d、e处原DAS 苯环上的质子峰以及b、c处-NH-的质子峰均存在,a 处Das 上的-OH 质子峰消失,且在δ=2.81、2.74×10-6出现新的-CH2-CH2-上的质子峰,因此可以认为DAS⁃SA 被成功合成。
2.3 检测波长的选择
称取DAS 对照品适量,用甲醇稀释成浓度适宜的样品,作为供试液。以甲醇溶液作为空白溶剂,在200~600 nm 波长范围内进行紫外扫描,考察其紫外吸收情况,结果见图1。

图1 DAS 的紫外波长扫描
由图1 可知,DAS 在322 nm 处有最强吸收。因此,选择322 nm 作为DAS 的检测波长。
2.4 色谱条件
色谱柱:Waters Symmetry C18(250 mm×4.6 mm,5 μm);流动相:0.3%磷酸盐缓冲液⁃乙腈(70 ∶30);流速:1.0 mL·min-1;检测波长:322 nm;柱温:30 ℃;进样量:20 μL。
2.5 系统适用性试验
取DAS 对照品适量,用甲醇溶解后稀释至适宜浓度,得到DAS 对照品溶液,将DAS 对照品溶液注入高效液相色谱仪,按照2.2 项下色谱条件进样,记录色谱图,见图2。
如图2 所示,在2.2 的色谱条件下,DAS 色谱峰呈正态分布且有较好的对称性,并且理论塔板数为5 289>3 000、分离度为1.857,>1.5,符合药典规定。
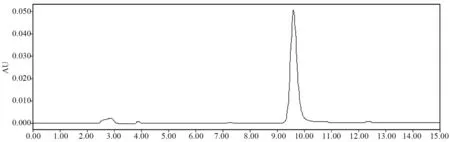
图2 DAS 的高效液相色谱图
2.6 专属性试验
按上述反应条件下的比例取DAS(反应物),SA(反应物)以及三乙胺(反应催化剂),加入适量DMF(反应溶剂)溶解后,用甲醇稀释至适宜浓度,制备混合对照品溶液。按照上述混合对照品溶液的制备方法,制备不含DAS 的阴性对照品溶液。另取合成产物适量加入少量DMSO 溶解后,用甲醇稀释成适宜浓度的样品得到DAS⁃SA 溶液。将DAS 对照品溶液、混合对照品溶液、阴性对照品溶液及DAS⁃SA 溶液分别注入高效液相色谱仪,按照2.2 项下色谱条件进样,记录色谱图。见图3、图4、图5。

图3 阴性对照品溶液的高效液相色谱图

图4 混合对照品溶液的高效液相色谱图

图5 DAS⁃SA 的HPLC 图谱
阴性对照色谱与对照品溶液色谱相同保留时间处无干扰峰,且混合对照品及DAS⁃AS 色谱图中DAS 峰与其他杂质峰及DAS⁃SA 的色谱峰均分离较好,表明方法专属性强,能满足检测要求。
2.7 线性关系考察
精密称取DAS 对照品10 mg,置于100 mL 容量瓶中,先用少量甲醇溶解,再用甲醇定量稀释至刻度线,摇匀,即得100 μg·mL-1DAS 对照品贮备液。用移液管精密量取10 mL 的100 μg·mL-1DAS 对照品溶液,置于50 mL 容量瓶中,甲醇定容至刻度,得到20 μg·mL-1DAS 对照品溶液作为母液。再将母液分别稀释成浓度为0.5、1.0、5.0、10、15、20 μg·mL-1的溶液,按照2.2 项下的色谱条件进样,记录相应的峰面积。以进样浓度(X)为横坐标,DAS的峰面积(Y)为纵坐标进行线性回归,得到线性回归方程。
以色谱峰面积为纵坐标Y,DAS 浓度为横坐标,绘制标准曲线。再通过峰面积Y对浓度X进行最小二乘法线性回归,DAS 的线性回归方程为Y=89 944X⁃4 256.3(r2=0.999 8),线性范围为0.5~20 μg·mL-1。结果表明:在线性范围内DAS 的色谱峰面积与浓度具有良好的线性关系。
2.8 精密度实验
配制浓度为15 μg·mL-1的DAS 对照品溶液6等份,分别在日内及日间对其重复测定,考察精密度,然后记录所测得的色谱峰面积,并通过2.4 项下所得的线性方程计算浓度,最后计算出其日内和日间的RSD 值。
精密度试验结果见表1。其精密度RSD 小于2%,精密度良好,符合《中国药典》2015 版的规定。

表1 精密度实验结果(n=6)
2.9 稳定性试验
分别取浓度为10 μg·mL-1、15 μg·mL-1、20 μg·mL-1的DAS 对照品溶液,分别在室温下放置0、1、2、4、8、12 h 后,测定其相应的色谱峰面积,将结果代入线性回归方程中,计算其RSD值。
通过对不同浓度的DAS 溶液在1 h、2 h、4 h、6 h、8 h、12 h 不同时间内稳定性的考察,其RSD<2%,结果表明其稳定性较好,见表2。

表2 稳定性试验结果(n=3)
2.10 方法回收率实验
分别取低、中、高三种不同浓度的DAS 对照品溶液,测定其相应的色谱峰面积,带入线性回归方程中,计算DAS 浓度,最后计算回收率及RSD 值。
对1、10.0、20.0 μg·mL-1三个DAS 溶液的浓度进行回收率的测定,测定结果见表3。回收率在99.66%~100.78%之间,RSD 值均小于2%,其回收率符合《中国药典》的98%~102%之间的规定,符合药典要求。

表3 方法回收率试验结果(n=3)
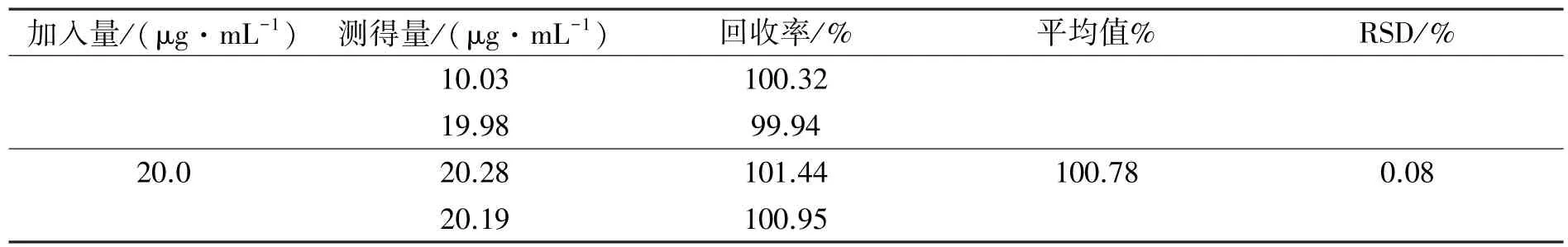
续表3
2.11 合成产物中DAS⁃SA 的含量测定
分别取三批合成产物适量,加入少量DMSO溶解后,用甲醇稀释成适宜浓度的样品,按照2.2项下色谱条件进样,记录色谱图,并根据回归方程计算各样品中DAS⁃SA 的相对含量,测定结果见表4。

表4 产率测定结果(n=3)
由图6 可知,DAS 的保留时间为9.651,其衍生物的保留时间为13.397,分离度较好。通过面积归一化法,即〔DAS⁃SA 的峰面积/除溶剂峰外所有的峰面积之和〕×100%计算,可得出最终产物的纯度,再通过产率=实际产物量×产物纯度/理论产物量,可得出该反应产率约为71.16%。
3 讨论
本研究利用DAS 上的羟基与SA 开环后的羧基进行酯化反应,通过核磁共振氢谱图证明了该DAS衍生物被成功合成,然后利用紫外分光光度计对DAS 进行全波长扫描,确定其最大吸收峰位于322 nm。最后建立高效液相色谱法(HPLC)来测定产物中的DAS 衍生物中DAS 的百分含量,并对该反应产率进行计算。结果表明,在该色谱条件下,DAS在0.5~20 μg·mL-1线性范围内色谱峰面积与浓度具有良好的线性关系(r2=0.999 8),且精密度、稳定性、方法回收率均符合药典规定。最终通过色谱峰面积比值得出在本文的反应条件下DAS 衍生物的纯度,并通过公式计算得出该反应产率为71.16%,反应产率较高。此外,利用高效液相色谱法对DAS衍生物进行定量分析具有快速高效、重现性好等优点,为DAS 衍生物的含量测定提供方法依据,也为进一步修饰DAS 以改善其理化性质和成药性奠定基础。
猜你喜欢
可再生能源(2022年5期)2022-06-09
今日农药(2017年10期)2017-11-14
科学家(2016年17期)2017-10-17
山东工业技术(2017年12期)2017-07-06
中学生数理化·高二版(2016年3期)2016-12-26
中学生数理化·高二版(2016年3期)2016-12-26
科技视界(2016年22期)2016-10-18
数理化学习·高三版(2009年3期)2009-04-30
中学生数理化·高二版(2008年2期)2008-10-19
