
法匹拉韦的合成工艺研究
2021-09-04 07:21魏天航徐明杰郭建超甲宗青苗雪凤王西龙山东道箴医药科技有限公司山东潍坊6000山东道一医药科技有限公司山东东营57000
中南药学 2021年8期
魏天航,徐明杰,郭建超,甲宗青,苗雪凤,王西龙*(.山东道箴医药科技有限公司,山东 潍坊6000;.山东道一医药科技有限公司,山东 东营 57000)
法匹拉韦(favipiravir,见图1)是一种新型RNA 依赖的RNA 聚合酶(RdRp)抑制剂,属于广谱抗流感病毒药物[1-3],由富士胶片集团富山化学工业株式会社开发,于2014年3月在日本批准上市[4]。除流感病毒外,其还对多种RNA病毒表现出良好的抗病毒作用,如埃博拉病毒、沙粒病毒、狂犬病毒等[5-7]。
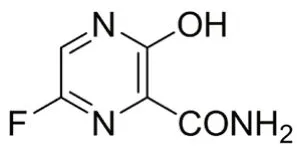
图1 法匹拉韦结构式Fig 1 Structure of favipiravir
本文在查阅相关文献后,对法匹拉韦的合成工艺进行了研究与改进,以期获得更加适合工业化生产的合成工艺路线。文献报道的法匹拉韦的合成路线主要有以下三种:
① 以3-羟基吡嗪-2-酰胺为起始原料,经硝化、氯代、氟代、水解、氧化等五步反应得到目标产物[8],如图2所示,该路线起始原料价格较贵不易得,路线中氯代试剂采用三氯氧磷,淬灭处理时与水剧烈反应,极易发生安全事故;工艺采用双氧水进行氧化反应,双氧水遇铁极易分解爆炸,对反应设备及储存设备要求较为苛刻;该路线中氯代及氟代中间体对人体皮肤具有极强的灼伤性,极大地限制了该工艺路线的放大生产。
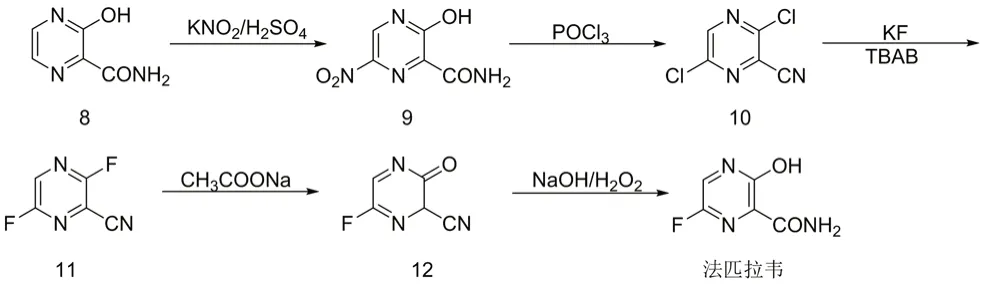
图2 法匹拉韦的合成路线1Fig 2 Synthetic route 1 of favipiravir
② 以2-氨基丙二酰胺为起始原料,如图3所示,经环合、溴代、氯代、氟代、水解、浓硫酸水解酰胺等六步反应得到目标产物[9-10],该路线起始原料廉价易得,但氯代试剂仍选用三氯氧磷,后处理存在安全隐患;该路线依然没有避开具有灼伤性的氯代及氟代中间体,限制了工业化生产。
③ 以3-氨基吡嗪-2-羧酸为起始原料,如图4所示,经酯化、溴代、重氮化引入甲氧基、Buchwald-Hartwing 反应、氨酯交换、重氮化、三甲基氯硅烷催化水解等七步反应得到目标产物[11-12],该工艺路线冗长,收率较低,且两次用到重氮化反应,在Buchwald-Hartwing 反应时用到昂贵的有机钯为催化剂且无法回收套用,成本较高。

图4 法匹拉韦的合成路线3Fig 4 Synthetic route 3 of favipiravir
为克服上述路线中的缺点,本研究对法匹拉韦的合成工艺路线进行了改进。本文以廉价易得的丙二酸二乙酯(2)为起始原料,如图5所示,经溴代反应得到2-溴丙二酸二乙酯(3),与氨气进行氨酯交换、取代反应得到2-氨基丙二酰胺(4),碱性条件下与乙二醛扣环得到3-羟基吡嗪-2-酰胺的钠盐(5),再经混酸硝化、Pd/C 催化氢化,最后经重氮化引入氟等六步反应得到目标产物法匹拉韦。

图5 法匹拉韦的合成路线Fig 5 Synthetic route of favipiravir
1 材料
HPLC 谱采用U3000 高效液相色谱仪(美国赛默飞,Chromeleon 7 色谱工作站)测定;NMR谱采用AVANCE NEO 400 核磁共振分析仪(瑞士Bruker)测定;ESI-MS 谱采用 Q Exactive 质谱仪(Q Exactive Plus LCMS,美国赛默飞)测定;JE1002 型电子天平(上海恒勤仪器设备有限公司);薄层层析板用 GF-254 型荧光板(上海旭泊实业有限公司);合成所用原料及试剂均为化学纯或分析纯,HPLC 所用试剂为色谱纯,起始原料丙二酸二乙酯(纯度:99%,上海润泰医药科技有限公司)。
2 方法
2.1 2-溴丙二酸二乙酯的制备(3)
将400.0 g 丙二酸二乙酯和600 mL 四氯化碳加到5000 mL 四口烧瓶中,缓慢升温至70 ~75℃,打开紫外灯,控制温度在70 ~75℃,通过恒压滴液漏斗缓慢滴加溴素的四氯化碳溶液(410.0 g溴素溶解在1600 mL 四氯化碳中),滴加速度根据反应瓶中红色褪去速度来确定,滴加结束后,在70 ~75℃下继续搅拌5 h,取样通过TLC 确认反应结束,降温至20 ~25℃,加入1000 mL5%碳酸钠水溶液,搅拌20 min,静置分层,有机相减压浓缩至无溶剂蒸出。
将上述残留液体转移至1000 mL 精馏塔中,减压精馏,得到547.4 g 无色油状产品,收率为92.0%。ESI-MSm/z:240.0 [M +H]+。1H-NMR(400 MHz,DMSO-d6)δ:1.20 ~1.24(m,6H,-CH3),4.19 ~4.25(m,4H,-CH2-),5.55(s,1H,-CH-)。
2.2 2-氨基丙二酰胺的制备(4)
将700.0 g 无水乙醇加到2000 mL 四口烧瓶中,缓慢降温并控制在0 ~10℃,将160 g 氨气慢慢通至四口烧瓶中,配制成液氨的乙醇溶液,控制温度在0 ~10℃下,通过恒压滴液漏斗缓慢滴加中间体3 的乙醇溶液(226.5 g 中间体3 溶解在200 mL 无水乙醇中),滴加结束后,缓慢升温至30 ~35℃,搅拌反应20 h,取样通过TLC 确认反应结束,缓慢降温至0 ~5℃,搅拌1 ~2 h,过滤,得到黄色固体。
将上述黄色固体加到450 mL 工艺水中,在20 ~25℃下搅拌40 ~60 min,缓慢降温至0 ~5℃,搅拌1 ~2 h,过滤,并用110 mL 工艺水淋洗滤饼,将滤饼置于鼓风干燥箱中,于55℃下干燥,得到99.6 g 黄色固体,收率为90%。ESI-MSm/z:118.1 [M +H]+。1H-NMR(400 MHz,DMSO-d6)δ:2.15(s,2H,-NH2),3.75(s,1H,-CH-),7.25(s,2H,-CONH2),7.43(s,2H,-CONH2)。
2.3 3-羟基吡嗪-2-酰胺钠盐的制备(5)
将380 mL 工艺水和84.5 g 氢氧化钠加到2000 mL 四口烧瓶中,搅拌至固体全部溶解,降温并控制在-5 ~5℃,加入200.0 g 中间体4,搅拌20 ~30 min,缓慢滴加300.0 g 40%的乙二醛水溶液,搅拌40 ~60 min,缓慢升温至20 ~25℃,并搅拌反应3 h,取样通过TLC 确认反应结束,缓慢降温至-5 ~5℃,搅拌1 ~2 h,过滤,将滤饼置于鼓风干燥箱中,于55℃下干燥,得到298.0 g 产品,收率为92%。ESI-MSm/z:162.1 [M +H]+。1H-NMR(400 MHz,DMSO-d6)δ:7.16 ~7.17(d,J=4.0 Hz,1H,-CONH-),7.30(d,1H,Ar-H),7.85(d,1H,Ar-H),10.94 ~10.95(d,J=4.0 Hz,1H,-CONH-)。
2.4 3-羟基-6-硝基吡嗪-2-酰胺的制备(6)
将53.0 g 浓硫酸(质量分数98%)加到100 mL 四口烧瓶中,降温至-5 ~0 ℃,之后分批次加入10.0 g 中间体5,搅拌至全部溶解,控制温度在-5 ~0℃下,缓慢滴加4.76 g 浓硝酸(质量分数68%),滴加结束后,缓慢升温至20 ~25℃,搅拌2.0 ~3.0 h,取样通过TLC 确认反应结束,将反应液淬灭到70 mL 冰水中,在10 ~15℃下搅拌1.0 ~2.0 h,过滤,用20 mL水淋洗滤饼,得到湿固体滤饼,硫酸水溶液用于制备聚合硫酸铁。
在上述固体滤饼加到40 mL 甲醇中,升温至回流,在回流状态下搅拌2.0 ~3.0 h,缓慢降温至20 ~25℃,在搅拌1 ~2 h,过滤,将滤饼置于鼓风干燥箱中,于55 ℃下干燥,得到9.4 g黄色固体,收率为71%。ESI-MSm/z:183.1 [MH]-。1H-NMR(400 MHz,DMSO-d6)δ:8.06(s,1H,-CONH-),8.32(s,1H,-CONH-),8.97(s,1H,Ar-H)
2.5 6-氨基-3-羟基吡嗪-2-酰胺的制备(7)
将3.0 g 中间体6 和192 g 甲醇加到500 mL高压反应釜中,用氮气置换3 次,加入0.3 g 5%湿钯碳催化剂,依次用氮气置换3 次、用氢气置换3 次,调整温度至45 ~50℃、压力至30 ~40 psi,并在此温度和压力搅拌反应10 h,取样通过TLC 确认反应结束,过滤除去钯碳催化剂,浓缩滤液,得到黑色固体,钯碳催化剂可以回收套用。
将上述黑色固体加到30 mL 丙酮中,缓慢升温至回流,并在回流状态下搅拌1 ~2 h,缓慢降温至20 ~25℃,搅拌30 ~50 min,过滤,将滤饼置于真空干燥箱中,于45 ℃下干燥,得到1.96 g 棕褐色固体,收率为78%。ESI-MSm/z:153.1[M-H]-。1H-NMR(400 MHz,DMSO-d6)δ:6.00(s,2H,Ar-NH2),7.55(s,1H,-CONH-),7.82(s,1H,-CONH-),8.48(s,1H,Ar-H)
2.6 法匹拉韦的制备(1)
在氮气保护下,将8.0 g 中间体7 和60 mL 70%氢氟酸吡啶溶液加到100 mL 四口烧瓶中,缓慢降温至-20 ~-15℃,在氮气保护下,加入7.5 g 亚硝酸钠,搅拌反应1 h;缓慢升温至20 ~25 ℃,搅拌2 ~3 h。反应结束后,缓慢加入24 mL 去离子水,用40 mL 醋酸异丙酯萃取,分层,有机相用16 mL 饱和食盐水洗涤,浓缩有机相,得到黄色固体。
将上述黄色固体加到30 mL 乙醇中,缓慢升温至回流,并在回流状态下搅拌1 ~2 h,缓慢降温至20 ~25℃,搅拌30 ~50 min,过滤,将滤饼置于真空干燥箱中,于55℃下干燥,得到5.3 g 白色固体,收率为65%,经HPLC 检测纯度99.96%, 单杂小于0.05%。ESI-MSm/z:156.0 [M-H]-;1H-NMR(400 MHz,DMSO-d6)δ:8.50~8.52(d,J=8.0 Hz,2H,-CONH2),8.75(s,1H,Ar-H),13.41(s,1H,Ar-OH)
3 结果与讨论
本文的起始原料廉价易得,第一步溴代反应生成的溴化氢经碱液吸收后生成溴化钠,处理后可额外获得经济效益;第二步氨气与酯发生氨酯交换,同时取代溴原子得到中间体4,反应结束后直接降温抽滤,操作简单,母液回收后固体残渣可进一步提取溴素,实现了闭环;第三步碱性条件下与乙二醛扣环,得到中间体5,反应完毕降温后直接抽滤即可,操作简便;第四步经混酸硝化,得到中间体6,淬灭打浆抽滤即可,母液经脱色后用于制备聚合硫酸铁;第五步氢化还原,催化剂采用Pd/C,反应清洁且后处理简单,回收后的Pd/C 可直接套用至下批反应,解决了贵金属钯成本高的问题;最后一步经重氮化引入氟,经纯化后HPLC 检测纯度可达99.96%,单个杂质均在0.05%以下。
本文工艺路线与现有报道合成路线相比,避免了三氯氧磷、双氧水等危险试剂的使用,反应条件温和、后处理简单,规避了强灼伤性氯代中间体的产生,工艺过程中产生的相关产物能够充分利用,进一步转化为经济效益,工艺总收率为27.5%,所得法匹拉韦纯度高、工艺经济、安全可行,工业化生产与现有工艺相比具有较大优势。
猜你喜欢
中国农业科技导报(2022年10期)2022-12-03
橡塑技术与装备(2022年8期)2022-08-05
中国药学药品知识仓库(2022年10期)2022-05-29
汕头大学学报(自然科学版)(2020年4期)2020-12-14
——非均布滤饼的局部比阻与平均比阻的测定与计算方法
化工装备技术(2020年4期)2020-09-09
流体机械(2020年5期)2020-06-24
石油钻探技术(2018年5期)2018-10-13
中国矿业(2017年2期)2017-02-28
股市动态分析(2015年12期)2015-09-10
邵阳学院学报(自然科学版)(2015年1期)2015-06-05
