
二维V型AlP半导体及其可调直接带隙
2021-09-02 09:24:48毛彩霞张玉萍杨旭鑫胡永红
华中师范大学学报(自然科学版) 2021年4期
毛彩霞,张玉萍,薛 丽,杨旭鑫,胡永红*,吴 涛
(1.湖北科技学院电子与信息工程学院,湖北 咸宁 437100;2.咸宁职业技术学院教务处,湖北 咸宁 437100)
石墨烯的发现为二维材料的发展打开了一扇大门,大量涌现的二维材料因具有优异的物理性质而备受关注,具有广泛的应用前景[1-3].已被理论预测和科学实验成功合成的二维材料在实际应用方面又有其自身的局限性,例如,石墨烯的零带隙限制了其在纳米电子器件中应用[4-6].因此,为了充分利用这些二维材料,人们采用了许多化学和物理方法来调控它们的性质,以满足各类新型纳米电子器件设计开发对半导体材料物理性质的需求.常用的方法主要有掺杂、化学修饰、施加应力和电场等[7-12].例如,石墨烯的带隙可通过外加应力来打开,因而扩展了石墨烯在场效应晶体管、太阳能电池和光电探测器等领域的应用.
AlP是元素周期表中典型的Ш-V族化合物,同时也是一种宽带隙半导体[13-14].由于其优异的电子和磁特性,使得块体AlP在光电探测器、发光二极管和自旋电子学等领域中具有重要的应用.人们用原子掺杂的方法来改变AlP的物理性质,并发现了许多新的电子和磁特性[15-19],这满足了新型纳米电子器件的要求.AlP在自然环境条件下具有闪锌矿结构,其实验晶格常数为0.546 nm,这与硅晶体的晶格常数非常接近.最近,在Ag(111)上进行了硅外延生长的实验,得到了二维硅烯.这自然而然地引发科学思考:是否存在可稳定存在的单分子层AlP或低维AlP纳米结构?如果存在,它们又会表现出什么样的物理特性?通过文献调研,发现有学者已经尝试回答了上述问题.例如,孙等人通过理论计算发现了单壁AlP纳米管的稳定性和电子结构.他们发现AlP之字形纳米管是直接带隙半导体,在太阳能光伏器件上有很大的应用潜力.通过对闪锌矿体晶格的剥离和优化,童等人从理论上得到大量闪锌矿化合物的二维单分子层材料,这其中包括二维四方型AlP(T-AlP),且它具有丰富的电子和光学特性.最近,刘等人基于密度泛函理论对单层二维T型AlP (T-AlP)进行了研究,发现单层T-AlP的电子能带结构特点可通过外加应力和范德华堆叠来调控.
本文基于密度泛函理论深入的计算研究了单分子层V型AlP(V-AlP)晶体的几何结构和电子能带结构特性.通过充分几何结构优化和声子谱计算分析证明V-AlP具有良好的结构稳定性.进一步研究其电子结构,发现它是一种宽直接带隙半导体材料(2.6 eV).然后详细考察了外加应力和电场对V-AlP几何结构和电子能带结构的影响.研究发现V-AlP的电子结构在外加应力和电场作用下具有丰富的变化行为,并且表现出对应力的各向异性.研究结果表明二维V-AlP晶体是未来纳米电子器件的重要候选材料.
1 方法和模型
本文的第一性原理计算研究是基于Materials Studio 8.0软件包中CASTEP模块的量子力学程序来实现的,采用Perdew-Burke-Ernzerhof (PBE)函数和Heyd-Scuseria-Ernzerhof (HSE)杂化泛函.对于HSE06函数,使用的屏蔽参数为2.1 nm-1,短距离交换的混合参数为0.25.垂直于二维面(沿c轴)方向取真空层厚度大于2 nm,以避免由相邻分子层之间范德华相互作用.考虑到层间的色散相互作用,基于Tkatchenko-Scheffler方法进行了范德华作用计算.在电子结构计算过程中,考虑Al原子的价电子态的基集为3s23p1,P原子的价电子态的基集为3s2p3.对于几何结构优化和电子能带性质计算,平面波截止能量设定为500 eV.分子结构充分弛豫,直到每个原子的净受力都小于0.1 eV·nm-1.同时还使用了15×15×1的Monkhorst-Pack-points网格,对二维布里渊区进行采样,用于结构优化和能带计算.凝聚能计算公式为Ecoh=ETot/N-γiEi,,其中ETot是系统的总能量,Ei是单个原子的能量.N是单位晶包内的原子数,γi是单位细胞中第i个原子的原子数与总原子数的比值.
2 结果与讨论
2.1 晶体结构和稳定性
图1为两种二维单分子层AlP超胞(T-AlP和V-AlP单层)结构优化之后的晶体结构图.厚度用符号Δ标记,对应结构的原胞单元用黄色矩形标记.红色和蓝色的球分别代Al原子和P原子.单层AlP的晶格参数、键长和厚度列于表1中.表1中,厚度由单分子层晶体材料的上下两层原子之间的垂直距离来定义.能量和长度的单位分别是eV和nm.括号中的值对应于HSE06方法计算的能量.图1(a)中V-AlP的晶体结构对称性属于空间群 PMN21,从俯视图来看,单分子层V-AlP是不等边的六边形环结构,垂直方向有较大的褶皱.Al (P)原子与三个相邻的P (Al)原子的键长不一样,沿晶格(a)方向的键长较短(0.229 nm),而另一个键长较长(0.232 nm).如图1(b)所示 T-AlP的四方结构属于空间群P4/NMM.它是层状结构,一个Al原子层夹在两个P原子层之间.每个Al (P)原子被其它四个P (Al)原子包围着.研究发现,V-AlP的平均键长(0.231 nm)比T-AlP (0.243 nm)的短4.9%,因此V-AlP比T-AlP稳定性更好.两种二维AlP模型的厚度不同(表1列出了它们的厚度值),单分子层V-AlP的厚度比T-AlP的薄23.4%.从表1可以看出,所得计算结果(晶格参数、键长和T-AlP单层厚度)与已有文献报道的数据高度吻合,可见所采用的计算方法是有效的,得到的晶体模型也是有效的.
稳定性的考察对于新型二维材料理论设计和实际应用都至关重要.为了考察两种二维AlP结构的动力学稳定性,进一步计算了它们的凝聚能.晶体的凝聚能普遍为负值,表明该材料具有良好的动力学稳定性.从表1的数据结果可以看出,两种类型的单层AlP均具有良好的稳定性(T-AlP和V-AlP的凝聚能分别为-5.35 eV和-5.12 eV),T-AlP的稳定性略优于V-AlP.通过外延生长得到的单层蓝磷晶体的结合能为-5.18 eV,与V-AlP单层的结合能基本相当.因此,可以在实验上制备单分子层V-AlP晶体材料.

图1 优化后的单分子层V-AlP(a)和T-AlP(b)单位超胞结构Fig.1 Optimized structures of V-AlP (a)and T-AlP (b)monolayers with the unit cells
如图2所示,V-AlP单分子层的声子色散谱中所有波矢量的声子谱中没有明显的虚模出现,因此判断V-AlP单分子层晶体的稳定性非常强.特别地,其最高频率高达535 cm-1,高于T-AlP (490 cm-1)[27]和MoS2单分子层(473 cm-1),表明在V-AlP单分子层晶体中Al和P之间有良好的键合.在谐波近似中,声子模的正频率和弹性稳定性准则是无应力晶体结构稳定的充分必要条件.二维V-AlP的弹性常数为C11=65.5 GPa,C12=8.4 GPa和C66=12.2 GPa,显然满足二维V-AlP晶格的力学稳定性标准(C11>C12和C66>0).
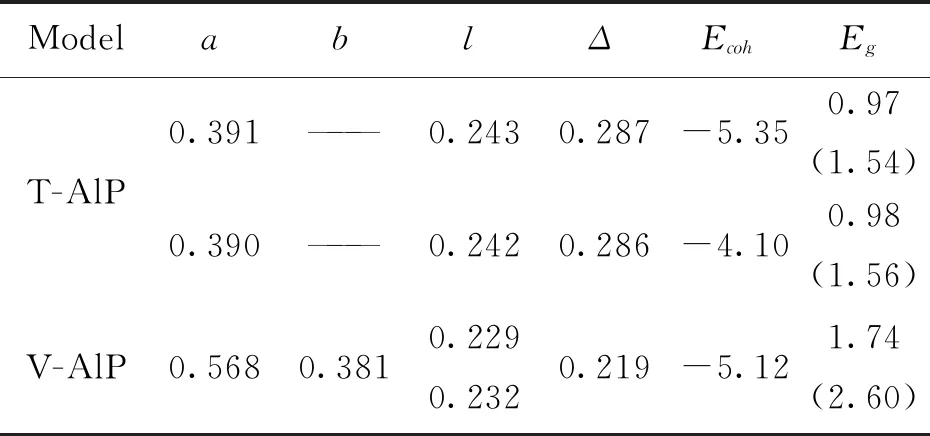
表1 单分子层AlP的晶格参数(a和b)、键长(l)、厚度(Δ)、凝聚能(Ecoh)和能量带隙(Eg).Tab.1 The lattice parameter (a and b),bond lengths (l),thickness (Δ),cohesive energy (Ecoh)and energy band gap (Eg) of the AlP monolayers
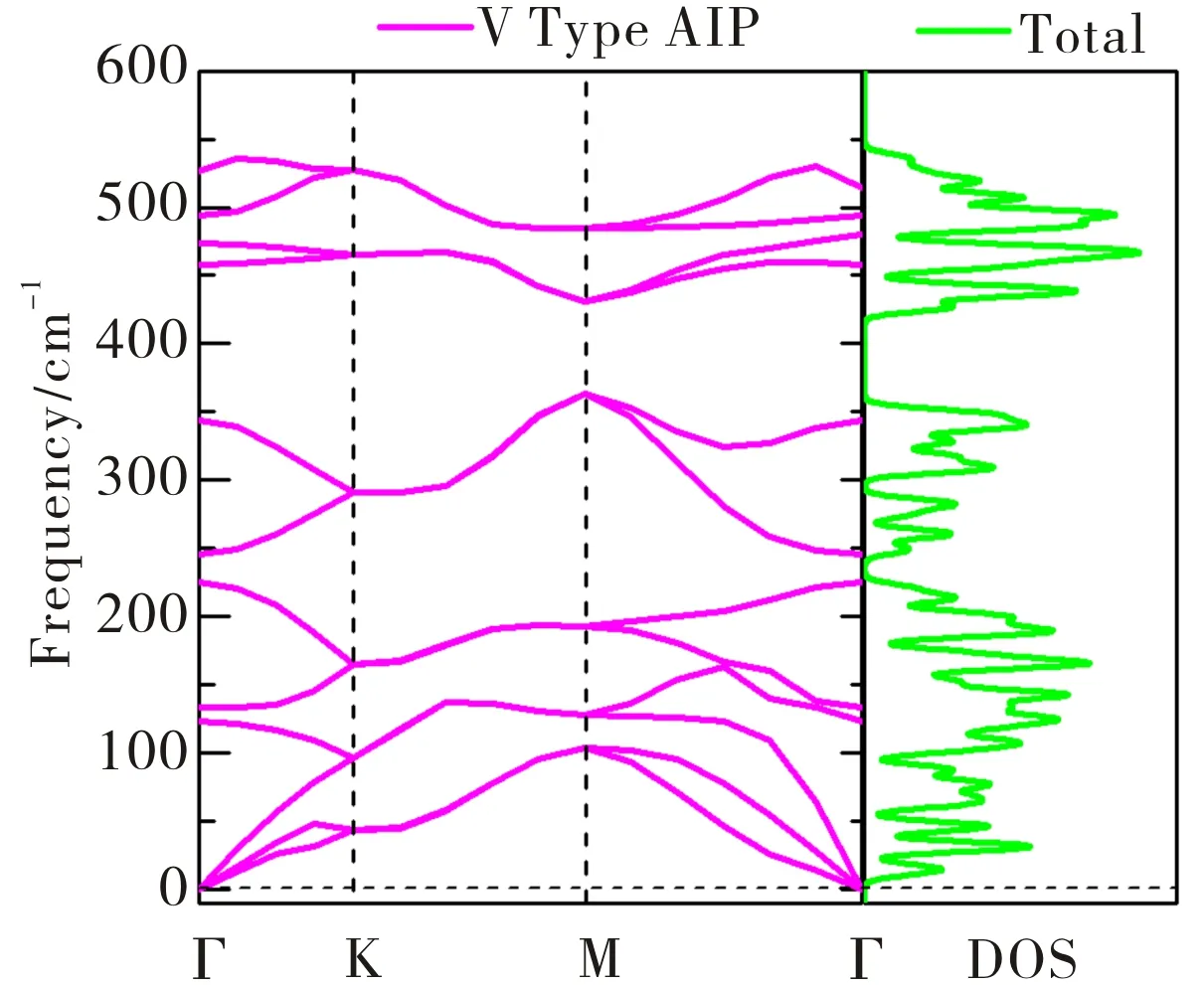
图2 计算得单分子层V-AlP原胞晶体的声子谱和相应的态密度Fig.2 The Phonon spectra calculated with the primitive cell of V-AlP monolayer and corresponding density of states
进一步,通过进行从头分子动力学(MD)模拟计算来考察外部温度对材料稳定性的影响.采用较大的初始原子超胞(共含64个原子)来增加相关长度.平面波截断能选取为500 eV,并选用了Norm-Conserving赝势来进行模拟计算.整个系统所处温度设定为T=973 K,模拟时间步长为1 fs,周期为1 ps.计算得系统总能量随MD模拟时间的变化情况如图3所示.图3中的分子结构图为分子动力学模拟1 ps结束时最终帧的快照.从图3可见,仿真结束时V-AlP晶体的几何结构基本保持不变,说明二维V-AlP晶体在高温下是稳定的.基于上述情况,T-AlP和V-AlP单分子层晶体有望在不久的将来被实验合成出来.
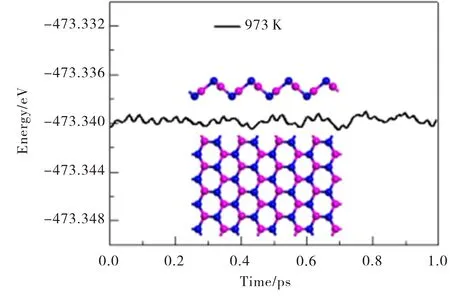
图3 在973 K下V-AlP单层超胞的总能量随分子动力学模拟时间的函数Fig.3 The total energy of the supercell of V-AlP monolayer under 973 K as function of MD simulation time
2.2 电子结构和光吸收系数
对于应用于纳米光电器件的2D半导体,直接带隙材料优于间接带隙材料.电子对光子的吸收需要间接带隙半导体中声子的参与,因此器件的光电转换效率收到限制.通常,可通过计算能带色散来深入了解V-AlP单层晶体的电子结构.图4显示了V-AlP单层的能带结构.如图4(a)所示,分别在PBE和HSE06水平获得1.74 eV和2.60 eV的直接带隙.如图4(b)所示,V-AlP的投影态密度(PDOS)表明P原子的P轨道主要贡献于价带边缘.能量范围从-5 eV到-3 eV时,P的p轨道与Al的p轨道杂化,导致电荷从Al转移到P,Al的s和p轨道与P原子的p轨道在费米能级以上杂化形成导带,能量范围为2 eV~4 eV.特别是Al和P的p轨道对导带底有积极的贡献.Al-P键的本质可以通过Hirshfeld电荷和重叠布局分析来说明.少量电荷(0.21|e|)从Al原子转移到P原子,加上Al-P键的重叠数(0.105),表明V-AlP是由Al-P共价键结合的.图4(c)显示了Γ点处的VBM和CBM,可以发现V结构中的轨道杂化既不是sp2也不是sp3.
基于吸收系数计算分析,研究了二维V-AlP在光照下的吸光性能.由偏振光计算出V-AlP纳米片的光吸收光谱.如图5所示,吸收光谱的峰分别位于5.00 eV和8.14eV左右.在3.28 eV到9.20 eV的光子能量范围内,吸收系数高于第一峰的三分之一(4.0×104cm-1),这意味着偏振光在相应的太阳光谱的几乎所有紫外线范围(134~378 nm)中显示出很强的光收集能力.因此,V-AlP表现出对紫外光的光响应,显示出在光电子器件中的潜在应用.

图4 单层V-AlP的能带结构(a)、态密度(b)和导带低和价带顶(c)Fig.4 Band structures(a),densities of states(b)and the minimum conduction band and the maximum valence band (c)of monolayer V-ACP

图5 基于HSE06杂化泛函计算的V-AlP单层的吸收系数Fig.5 Calculated absorption coefficient for V-AlP monolayer at the HSE06 level
2.3 应力和电场效应
应力是调控2D半导体材料能隙的一种有效的方法.应力可以被视为施加到2D材料上的弹性场,其通常改变材料的几何结构和电子性质.在一定的应力下,材料的几何结构由于弹性场和二维晶体场之间的相互作用而改变,然后材料的电子性质随着2D材料中电子密度的重新分布而改变.然而,一些典型的2D材料(如石墨烯)的能带结构对应力场并不敏感.因此,有必要考察外加应力是否能有效地调节V-AlP的电子结构特性.在图6中,在沿晶格方向施加的轴向应变下-20%至+8%,保持了V-AlP的直接带隙特性.直接带隙的值随着拉伸和压缩的增加而减小.随着双轴拉伸应变的增加,VBM从Γ点移动到近点,当拉伸从+8%增加到+10%时,发生直接-间接带隙转变.如图7所示,带隙跃迁源于近带边能态的竞争.间接带隙单调降低至+20%,没有任何间接-直接带隙跃迁.在HSE水平下,通过双轴应变获得的V-AlP直接带隙的可调谐范围为1.0 eV至2.6 eV.该范围覆盖了大部分可见太阳光能量范围(1.6 eV至3.1 eV).在图6中,V-AlP的带隙在单轴和双轴应变下的变化趋势呈现从-10%到+10%的比例.很明显,这三条带隙值的变化曲线是不同的.首先,不同应力条件下V-AlP的带隙都在单调增加,然后随着应变的增加而单调减少.然而,带隙曲线的弯曲点(或极值点)是不同的.这种应变效应的各向异性可以用特殊的V型结构来解释,它也具有各向异性的特性.太阳能电池材料的理想带隙是1.5 eV.V-AlP单层的直接带隙可以在很宽的范围内(从1.0 eV到2.6 eV)调谐,因此,它将在超薄光伏材料中有潜在的应用.研究结果表明,V-AlP单层的电子性质对轴向和双轴应变高度敏感.因此,V-AlP带隙的重要变化可以在设计柔性电子学和光电可调谐光电探测器中得到广泛应用.

图6 基于HSE06泛函计算的V-AlP带隙随施加单轴和双轴应力的变化Fig.6 Changes in bandgap with applied mono-axial and biaxial strain for V-AlP calculated by HSE06 functional
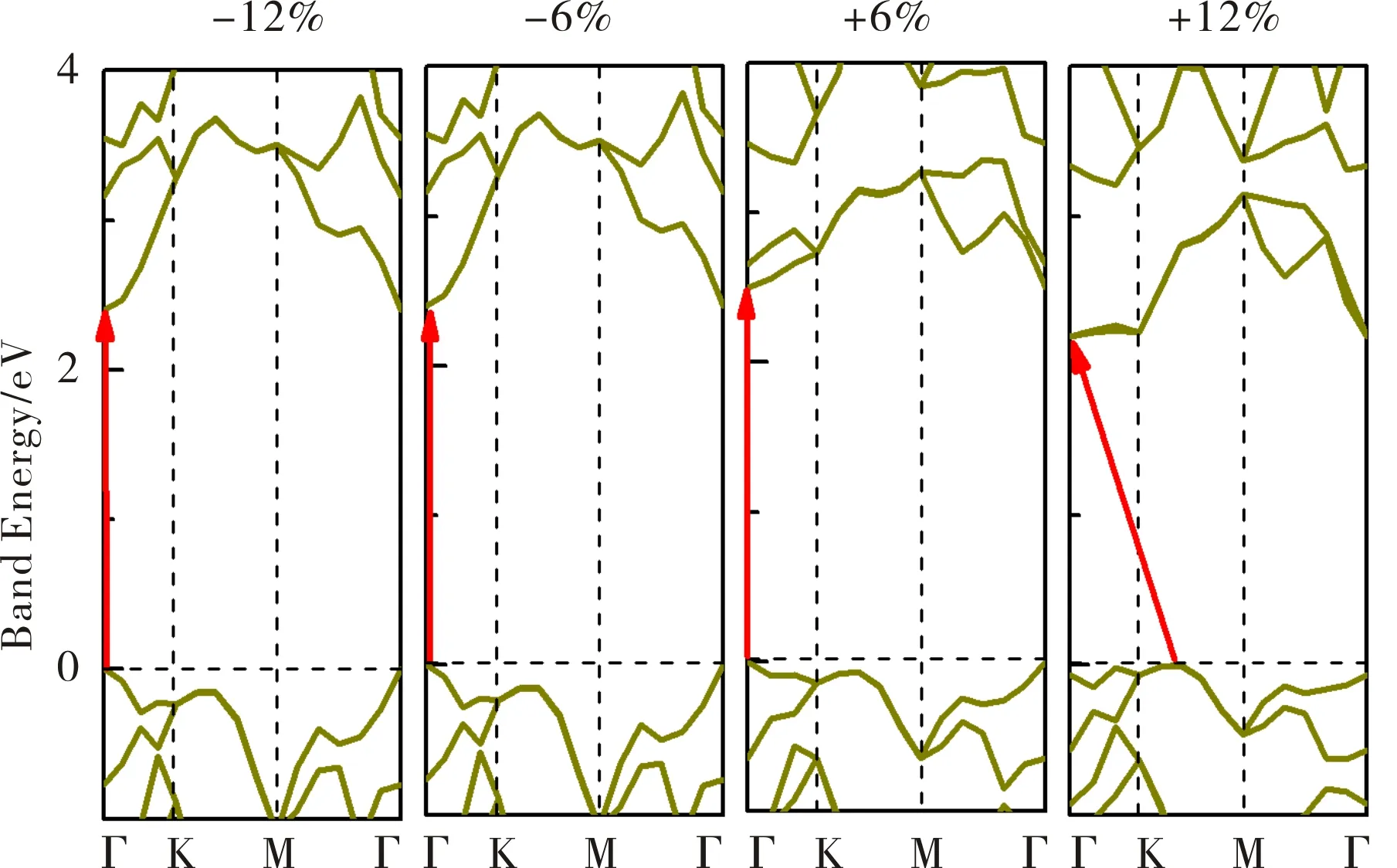
图7 基于HSE泛函下,V-AlP在双轴应力下的能带结构变化和带边移动Fig.7 Band structure change and the band edge shift under biaxial strains for V-AlP based on HSE functional
外加电场也是另一种常用的调控二维材料电子能带结构的有效方法.因此,在此接着考察在垂直于V-AlP平面方向上施加静电场对其电子能带结构的影响.图8中绘出了V-AlP单层的带隙随外电场的变化情况.从图8可见,V-AlP单层晶体的带隙值随电场强度的变化趋势可以分为两个区间来讨论.当沿Z方向的外电场低于5 V·nm-1时,随着电场强度的增加,带隙缓慢单调减小,下降斜率近似为-0.003 eV·nm·V-1.当外电场超过5 V·nm-1时,带隙快速单调减小,下降斜率近似为-0.65 eV·nm·V-1,与前者的比值约为217.而且,当电场强度超过9 V·nm-1时,也能观察到金属性质.如图9所示,当外部电场增加到9.5 V·nm-1时,能隙闭合,表明由于VBM的上移,从半导体性质到金属性变化.因此,V-AlP单层晶体的电学性质可以在直接带隙半导体和金属之间进行调控.这里,所有这些结果都可以由内置电场和外加电场之间的相互作用来解释[12].也就是说,当外加电场较弱时,总电场增加,带隙缓慢减小.当总电场达到一定值时,外部电场的作用大大增加,然后外部电场强度的进一步增加会导致带隙迅速减小.另外,还考虑了外加电场作用下V-AlP单层晶体中键间Hirshfeld电荷转移的变化.当开始施加外部电场时,Hirshfeld电荷转移不变(每对Al-P键为0.48|e|).当电场超过5 V·nm-1时,Hirshfeld电荷转移改变(每对Al-P键0.50|e|),这意味着外部电场也会影响电子密度的分布,导致带隙迅速减小.为了进一步探讨外加电场下带隙变化的机理,又研究了不同外加电场下的带边变化.图9展示了在某些外部电场下的带边缘,可以看出,随着外加电场的增加,V-AlP单层的CBM线性下降.因此,带隙减小主要是由于CBM的向下转移.幸运的是,研究发现在沿z方向的外电场下的V-AlP单层晶体保持直接带隙,并且直接带隙可以在较宽的范围内(从0到2.6 eV)被外加电场线性地调控.这种大尺度的带隙线性调控表明沿z方向施加外电场可以有效地动态控控基于V-AlP单层晶体的电子纳米器件的能带结构和电子性质.

图8 基于HSE泛函下,带隙值随垂直于V-AlP单层平面的z方向电场的变化Fig.8 The band gap as a function of E-field along the z direction perpendicular the plain of the V-AlP monolayer based on HSE functional

图9 沿几种z方向电场下V-AlP单层晶体的能带结构Fig.9 The band structures of V-AlP monolayer under certain E-field along the z direction
3 结论
本文基于密度泛函理论计算,系统地研究了单分子层V-AlP晶体的几何结构特点、稳定性和电子能带结构.V-AlP单层晶体比T-AlP单层晶体厚.负结合能、分子动力学模拟和声子谱的计算结果证明了V-AlP单层晶体的良好稳定性.另外,V-AlP单层晶体具有2.6 eV的宽直接带隙,因此在光电子器件中有潜在的应用.二维V-AlP单层晶体的带隙容易受外加应力和电场的调控.在外加双轴应力下(从-20%至20%),V-AlP的直接带隙可从1.0 eV调增到2.6 eV.V-AlP单层晶体的电子能带结构还表现出了对应力的各向异性特点.同时,在外加垂直(沿z方向)电场(从5 V·nm-1到10 V·nm-1)作用下,V-AlP的直接带隙也可以在很宽的范围内(0 eV到2.6 eV)线性调控.因此,外电场可以用来有效地改变二维V-AlP晶体的电子结构.这些研究结果为基于为V-AlP材料的纳米电子器件的设计提供了重要理论参考.
猜你喜欢
分子催化(2022年1期)2022-11-02 07:10:16
济南大学学报(自然科学版)(2021年6期)2021-11-10 01:25:42
小天使·聪聪画刊(2021年2期)2021-09-10 07:22:44
汽车零部件(2020年10期)2020-11-09 03:41:42
成都信息工程大学学报(2019年3期)2019-09-25 08:31:14
汉语世界(The World of Chinese)(2019年6期)2019-09-10 07:22:44
电子制作(2019年15期)2019-08-27 01:12:04
测控技术(2018年9期)2018-11-25 07:44:44
振动工程学报(2017年4期)2018-05-31 12:38:00
电子制作(2018年1期)2018-04-04 01:48:38
