
高效液相色谱-串联质谱法测定马铃薯中的α-茄碱和α-卡茄碱
2021-09-01 12:35:24陈琳珊陈佩佩吴永坤
现代食品 2021年14期
◎ 陈琳珊,陈佩佩,陈 茹,吴永坤
(1.广东省食品工业公共实验室,广东 广州 511442;2.广东省食品工业研究所有限公司,广东 广州 511442;3.广东省质量监督食品检验站,广东 广州 511442;4.中山市小榄镇农产品质量检验检测中心,广东 中山 528400)
茄碱是一种有害的生物碱类物质,主要以糖苷的形式存在,一般出现在茄科类作物中,尤其是腐败的茄科蔬菜[1],是导致茄科蔬菜中毒中的常见毒素之一。其主要成分为α-茄碱和α-卡茄碱,结构式见图1。茄碱抑制体内胆碱酯酶的活性,从而使乙酰胆碱积聚,引起神经兴奋,胃肠肌肉痉挛,中毒时出现腹痛、腹泻、恶心和呕吐等症状,情况严重时可导致脱水、发热、昏迷,甚至呼吸衰竭[2-3]。尽管有研究表明,茄碱对前列腺、肝、乳腺等癌细胞有一定的抑制作用[4-6],但对消费者日常饮食的危害也不容忽视。
图1 α-茄碱和α-卡茄碱的化学结构式图
目前,针对茄碱的分析方法主要分为化学方法、成像技术和生物技术3大类[7],由于分光光度法[8]、成像技术[9]、放射免疫[10]、酶联免疫[11]和比色法[12]等方法存在定量不准确、操作烦琐、耗时长和价格昂贵等缺点,科研工作者的研究主要集中在高效液相色谱法[13]和液相色谱-串联质谱法[14]。但高效液相色谱法需调节流动相使目标物有效分离,因此分析时间长,且α-茄碱和α-卡茄碱吸光度较弱,因此检测波长较低,容易受杂质干扰,影响分析结果。质谱法具有定性定量能力强、分析时间短、灵敏度高等优点,适合常规实验室的批量化检测。本文建立的高效液相色谱-串联质谱法,经方法学验证,具有较高灵敏度,同时回收率高,重现性好,可为后续研究提供参考。
1 材料与方法
1.1 仪器与试剂
API4000Q TRAP质谱仪(美国AB SCIEX公司)、岛津LC-20AD液相色谱系统(日本岛津公司)、4k-15离心机(北京卓明贸易有限公司)、IKA Vortex4涡旋混匀器(广州仪科实验室技术有限公司)、Milli-Q Advantage A10 超纯水系统(法国Merck Millipore公司)。
α-茄碱、α-卡茄碱(纯度大于98.5%,德国Dr.Ehrenstorfer公司,购自上海安谱公司)、甲酸、甲醇、乙腈(LC-MS级,美国Thermo Fisher公司)、C18、PSA、GCB、硅胶、NH2粉、中性氧化铝、碱性氧化铝(分析纯,购自上海安谱公司)。
1.2 标准溶液的配制
标准溶液:分别称取两种茄碱适量标准品,用甲醇溶解并稀释成浓度为1 mg·mL-1的标准溶液,于4 ℃下避光保存。
混合标准工作溶液:将标准溶液用乙腈稀释为10 mg·L-1的中间液,将马铃薯基质溶液依次稀释为10 μg·L-1、20 μg·L-1、50 μg·L-1、100 μg·L-1、200 μg·L-1、500 μg·L-1、1 000 μg·L-1制作校准曲线,标准加入法定量。
1.3 样品前处理
马铃薯样品均质。称取2.5 g样品于50 mL离心管中,加5 mL 水和20 mL 0.5%甲酸乙腈溶液(V/V),涡旋混匀,加4.0 g无水硫酸镁和1.5 g醋酸钠粉末,涡旋提取10 min。后置于离心机中,以10 000 r·min-1离心5 min,上清液转移至25 mL比色管中,并定容混匀。取1 mL提取液用50 mg C18净化,过膜上机。
1.4 色谱条件
色谱柱:Xbridge BEH C18(100 mm×2.1 mm,1.7 μm);进样量:10 μL;柱温:35 ℃;流动相A(乙腈)-B(0.1%甲酸水溶液)(V/V);流速:0.35 mL·min-1;梯度洗脱程序:0.0~0.5 min:40%A,0.5~3.0 min:40%~90%A,3.0~3.5 min:90%A,3.5~4.0 min:90%~40%A,4.0~5.0 min:40%A。
1.5 质谱条件
离子源电离方式:正模式;气帘气压力:25 psi;电离电压(IS):5 500 V;离子源温度(TEM):550 ℃;雾化气(GAS1):50 psi;加热辅助气(GAS2):50 psi;碰撞气(CAD):medium;采集模式:多反应监测(MRM)模式。
2 结果与分析
2.1 质谱条件的确定
α-茄碱和α-卡茄碱均为糖苷,由于其主体结构上含有一个叔胺碱的N原子,在ESI源正负离子模式均有响应。经两种模式对相应的母离子扫描后发现,正离子模式下母离子[M+H]+比负离子模式下的[M-H]-的响应更强,可能是因为主体结构上的N原子或糖苷上的O原子获取H原子的能力比糖苷上羟基-OH失去H原子更容易发生。确定母离子后,通过调节碰撞电压(Collision Energy,CE)获得尽可能多的二级子离子,以多反应监测模式分别对去簇电压(Declustering Potential,DP)及子离子对应的碰撞电压进行优化,α-茄碱和α-卡茄碱的MS/MS参数见表1。
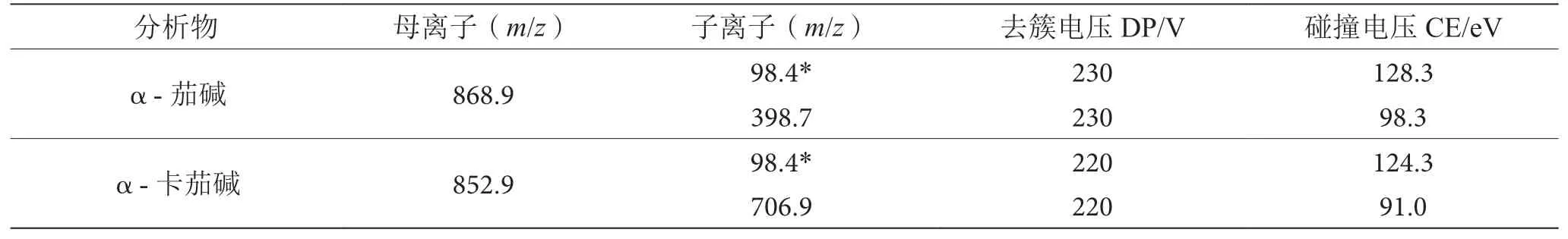
表1 多反应监测方法测定α-茄碱和α-卡茄碱的MS/MS参数表
2.2 色谱柱的选择
α-茄碱和α-卡茄碱结构十分相似,其差异仅为糖苷位置不同和糖苷上的羟基数量不同,在化学式上的差异也仅有一个O原子的质量数,因此其色谱行为也十分相似,在色谱中难以分离,尽管对于三重四极杆结构来说,可不作细致的色谱分离,但由于化学式相似,为避免α-茄碱在离子源内裂解出m/z852.9,影响定量结果,实验使用了安捷伦、Waters和菲罗门公司的3款色谱柱,对2种目标物进行分离。如图2所示,只有Waters公司的Xbridge BEH C18色谱柱有良好分离效果,原因可能是色谱柱的粒径大小对分离效果有较大影响,因此选择Xbridge BEH C18色谱柱。
2.3 固相萃取剂种类的选择
在实验初期发现,在乙腈中加入甲酸能提高回收率,但体积在20 mL以上时,回收率变化不大,因此在此基础上对固相萃取剂种类进行探究。使用量为50 mg时,结果显示C18的回收率最高,GCB和碱性氧化铝对目标物吸附严重,回收率极低。尽管PSA和硅胶能吸附糖类或其他大极性杂质,但回收率也低于50%,综合考虑,选择C18对提取液进行净化,详细结果见图3。
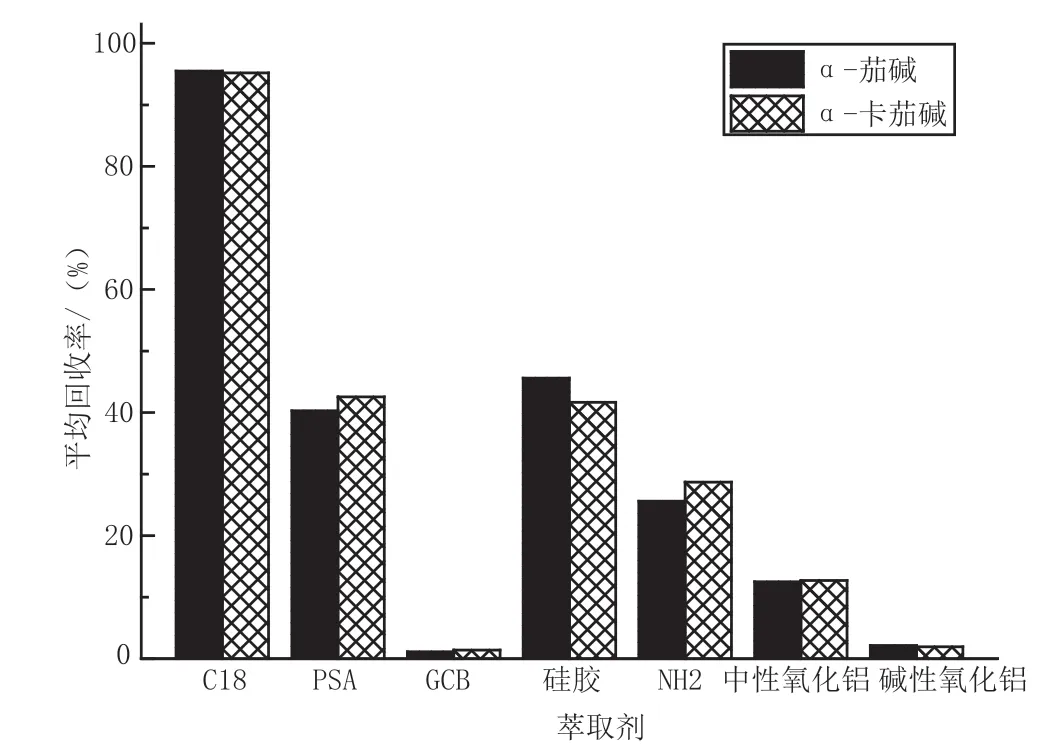
图3 固相萃取剂种类对回收率的影响图
2.4 基质效应
样品含有淀粉较多,若使用针对淀粉进行净化的固相萃取剂,对目标物也有严重吸附,因此可能存在一定的基质效应。为进一步考察,实验使用乙腈溶液配制浓度为100 μg·L-1的单点标准溶液,取2份马铃薯基质液,1份直接测定本底值,1份用于配制基质单点,浓度为100 μg·L-1。以乙腈溶液配制的单点计算,扣除本底值后,基质单点中α-茄碱和α-卡茄碱的浓度分别为43.5 μg·L-1和48.1 μg·L-1,说明基质效应抑制响应值比例为原来的45%,小于80%,需用基质曲线进行校正,由于没有找到空白的基质样品,因此校正浓度截距后定量。
2.5 定量方程及准确性
使用马铃薯基质液配制曲线后测定,以峰面积(Y)对质量浓度(X)进行线性回归,得到的曲线的X轴截距的绝对值为该样品含量,若要使用此曲线对其他马铃薯样品进行定量,需将X轴截距校正至原点。在1.2的线性范围内(10~1 000 μg·L-1),测定α-茄碱和α-卡茄碱的曲线方程分别为Y=88.5X+3 800和Y=157X+2 970,相关系数R2为0.999 6和0.999 4。按照信噪比的3倍和10倍不断稀释样品浓度后,方法检出限均为0.1 mg·kg-1,定量限均为0.3 mg·kg-1。线性范围、相关系数、检出限和定量限均满足一般样品中α-茄碱和α-卡茄碱含量的测定,达到准确定量。
2.6 方法学结果评价
按1.3对样品进行测定,选择含量最低的样品平均测定6次,取其平均含量作为本底值。按照本底值附近的1倍、2倍和5倍3个水平进行加标。每个水平测定6次,α-茄碱和α-卡茄碱的3个添加水平的平均回收率范围在85.7%~99.5%,相对标准偏差RSD在1.89%~3.31%。回收率及精密度均能达到定量要求,结果见表2。
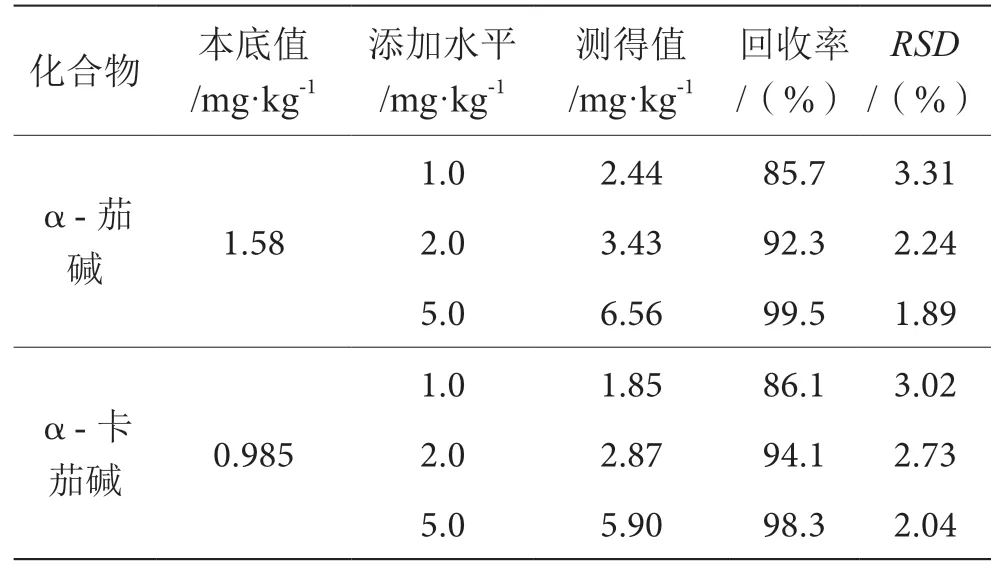
表2 α-茄碱和α-卡茄碱的回收率和精密度表(n=6)
3 结论
建立HPLC-MS/MS法同时测定马铃薯中α-茄碱和α-卡茄碱,使用甲酸-乙腈萃取后,盐析离心,进一步使用C18固相萃取剂净化,回收率达85%以上;多反应监测模式对母离子和子离子进行扫描,优化了质谱参数,使方法整体检出限和定量限达到分析定量要求;使用不同色谱柱进行对比,在优化的色谱条件下,α-茄碱和α-卡茄碱2 min内完全分离,且分离度高;3水平加标回收实验的回收率和相对标准偏差结果表明,该方法可作为日常马铃薯品质监控的有效手段。
猜你喜欢
煤化工(2022年3期)2022-07-08 07:24:42
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:06
中成药(2018年12期)2018-12-29 12:25:44
中成药(2017年6期)2017-06-13 07:30:35
食品界(2016年4期)2016-02-27 07:36:47
中国资源综合利用(2016年10期)2016-01-22 08:36:09
物理化学学报(2015年7期)2015-12-30 12:13:08
医学研究杂志(2015年4期)2015-06-10 06:42:43
应用化工(2014年3期)2014-08-16 13:23:50
中国洗涤用品工业(2011年3期)2011-03-20 15:38:06
