
系统性硬化症合并吉兰-巴雷综合征1例并文献复习
2021-08-31 01:45:08王浩玥沈雪阳何亚玲张正义
中风与神经疾病杂志 2021年7期
包 鑫, 王浩玥, 沈雪阳, 何亚玲, 张正义
系统性硬化症是一种临床表现多样,多器官受累的自身免疫性疾病,病理特点为皮肤和内脏的纤维化并伴有血管病变。吉兰-巴雷综合征是一种免疫介导的急性炎性多发性神经根神经病,多累及周围神经系统。本文报道1例系统性硬化症合并吉兰-巴雷综合征的诊治经过,并结合文献复习,对该类疾病的临床特点及诊治做一总结。
1 临床资料
患者,女,50岁。因“双手变白发紫1 y,面颈部皮肤紧绷伴双手肿胀0.5 y” 于2017年3月20日就诊于西安某医院,患者起初双手遇冷后出现皮肤变白发紫,后病情发展致面部、颈部、腹部皮肤紧绷,双手指肿胀。查体可见面颈、腹壁及双手皮肤紧绷、发硬,双手指肿胀,指腹末端有针尖大小皮肤破溃结痂。诊断“系统性硬化症”。给予醋酸泼尼松片60 mg起始治疗,同时予以阿司匹林肠溶片、贝前列腺素钠、雷贝拉唑、枸橼酸钾等对症治疗,后于2017年6月22日复诊,复查尿蛋白可疑,微循环中度异常,环磷酰胺位点检测毒性风险低,调整治疗方案“强的松30 mg减至20 mg/d,复方环磷酰胺片100 mg/d”维持治疗1 y余。2019年9月6日因“手指破溃1 m余”再次复诊,查免疫指标:补体C3、C4水平正常,ANCA靶抗原(MPO,PR3)阴性,肺功能:弥散功能轻度降低,心脏超声:肺动脉收缩压约22 mmHg,心包积液(少量),上消化道造影提示:食管蠕动减弱。给予对症治疗,激素逐渐减量至7.5 mg/d维持,皮肤僵硬症状明显改善。患者因“进行性肢体无力6 d” 于2020年7月9日就诊于我院,查体:双手、双侧腕关节以下皮肤增厚、紧硬、不易提起,双手皮温低,双上肢、双下肢肌力1/5级,肌张力减弱,腱反射消失,深浅感觉对称存在。辅助检查:脑脊液蛋白细胞分离(蛋白2.64 g/L,白细胞2×106/L)。免疫球蛋白:C3 0.93 g/L,C4 0.25 g/L,IgG10.63 g/L,IgA1.94 g/L,IgM1.66 g/L,抗核抗体阳性(滴度1∶100)。ANA谱(印迹法):Ro-52阳性,AMA M2阳性,Scl-70阴性,PM-Scl弱阳性,SS-A弱阳性,Jo-1阴性,抗着丝点抗体CENPB阴性,SS-B阴性,nRNP/Sm阴性,Sm阴性,神经节苷脂抗体检测(BLOT):抗GQ1b抗体IgG/IgM、抗GT1b抗体IgG/IgM、抗GD1b抗体IgG/IgM、抗GD1a抗体IgG/IgM、抗GM1抗体IgG/IgM、抗GM2抗体IgG/IgM、抗GM3抗体IgG/IgM均为阴性。心电图提示完全性右束支传导阻滞,胸部平扫提示双肺间质性病变。肌电图:双正中神经、右尺神经、双腓肠神经、右腓浅神经感觉神经传导均正常。双正中神经、双尺神经、右腋神经、右腓总神经运动神经诱发电位波幅均降低。左腓总神经运动神经传导速度轻度减慢,诱发电位波幅降低,远端潜伏期尚可。双胫神经H反射均未引出肯定波形(见表1)。诊断:系统性硬化症合并吉兰-巴雷综合征。给予静脉注射丙种球蛋白(17.5 g/d)治疗5 d后,患者肢体肌力2/5级,腱反射仍消失,无呼吸肌受累临床表现。继续给予醋酸泼尼松7.5 mg治疗系统性硬化症,转康复医院行肢体功能康复训练1 m余。5 m后患者四肢肌力3/5级左右,仍不能独立行走,生活难以自理。
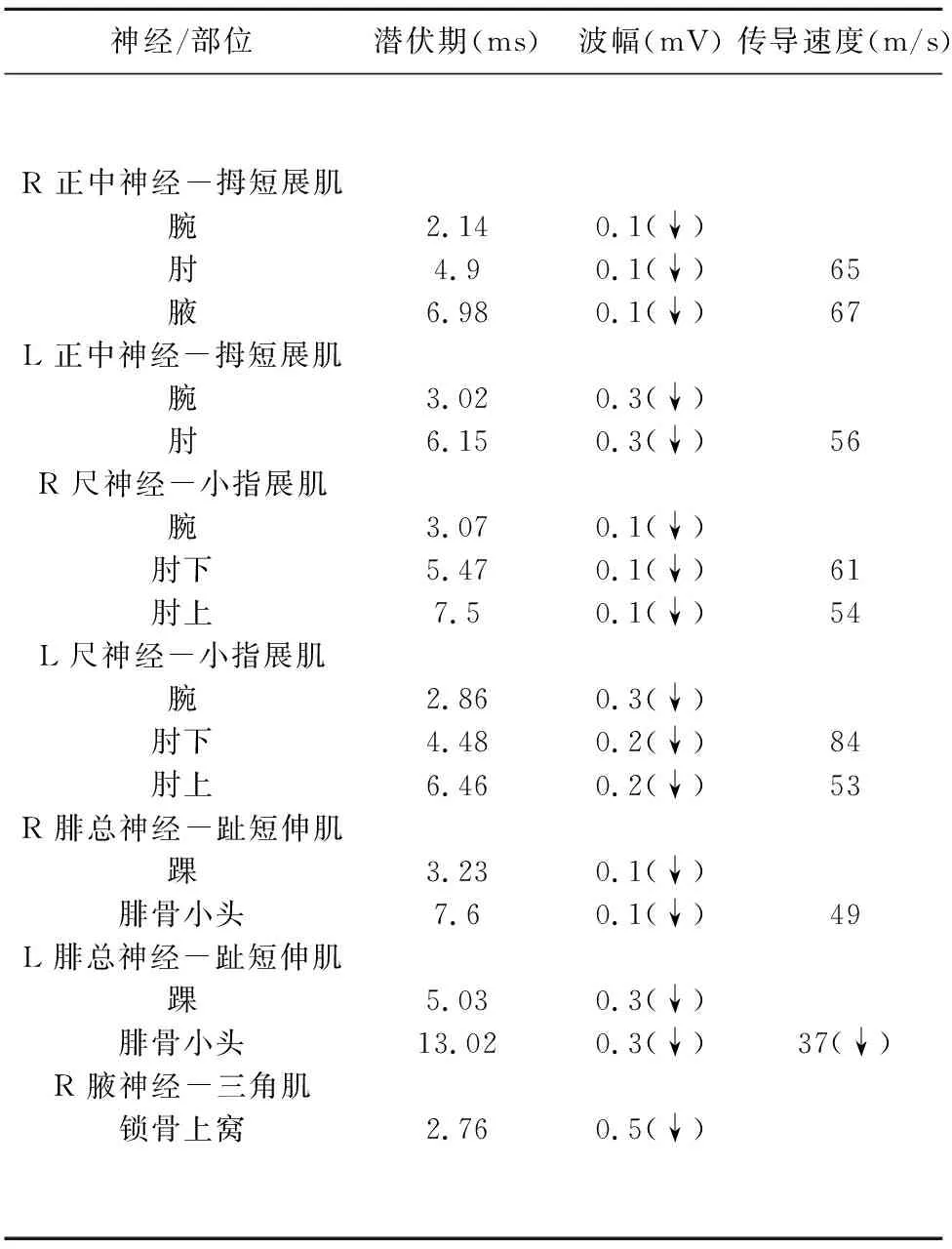
表1 发病7 d肌电图结果
2 文献复习
目前关于系统性硬化症合并吉兰-巴雷综合征的报道较少。查阅国内外中英文文献数据库,共检索到2例系统性硬化症合并吉兰-巴雷综合征。Yamaguchi等[1]报道了1例71岁老年男性患者,系统性硬化症病史4 y,规范口服泼尼松治疗,发生皮肤带状疱疹6 w后合并吉兰-巴雷综合征。查抗核抗体阳性,但抗Scl-70、抗着丝点和抗RNP抗体均为阴性。脑脊液检查显示蛋白细胞分离(蛋白0.94 g/L,白蛋白0.62 g/L,细胞数3×106/L)。神经传导检查显示运动传导速度缓慢、远端潜伏期延长和复合肌肉动作电位的时间离散。未检测到腓肠神经的感觉神经动作电位。抗GM1(80)、GD1b(80)和asialo-GM1(160)的血清IgM抗体为阳性。左侧腓肠神经活检提示:急性-慢性多发性神经病。给予甲泼尼龙注射液1g/d×3 d,随后口服泼尼松龙60 mg/d治疗。在住院13~17 d期间,共给予高剂量95gγ-球蛋白后,肌无力和感觉障碍均停止进展,然后逐渐改善。4 m后患者神经系统完全恢复,可以独立行走。
陈桂芳等[2]报告另外1例32岁女性患者,因“关节肿痛、双手遇冷变色1 y,肌无力4 d”就诊。入院后肌无力对称性进行性加重,以近端明显逐渐波及远端,10 d后四肢全瘫,双上肢肌力0级,双下肢肌力1级,肌张力低,腱反射消失。四肢远端手套、袜套样痛触觉减退。3次脑脊液均蛋白细胞分离(蛋白分别为1.4 g/L,0.98 g/L,3g/L,细胞数相应为4×106/L,3×106/L,5×106/L),肌电图第1次提示双上肢周围神经源性损害,第2次提示四肢周围神经源性损害,F波消失。予以泼尼松龙1g/d×3 d,后予以泼尼松60 mg/d维持治疗,于3 w病情稳定,4 w后神经系统症状逐渐恢复。
3 讨 论
系统性硬化症(systemic sclerosis,SSc)是一种临床罕见的结缔组织疾病,病理基础是血管损伤、炎症和结缔组织修复之间的相互作用。其特征是免疫功能紊乱,内皮细胞功能障碍,继而出现皮肤和内脏血管修复缺陷、新生血管和进行性的组织纤维化,但其详细的发病机制目前仍不清楚[3]。病变的神经内皮调控机制包括内皮细胞和周围神经纤维损伤,这被认为在雷诺现象的发生和微血管异常的发展中起着重要作用,而雷诺现象和微血管异常是SSc的最早病变和关键特征。这种致病的神经内皮机制可能会触发早期的内皮细胞损伤和随后的以缺乏代偿性血管生成为特征的外周微血管完整性的丧失。SSc具有遗传易感性,触发因素主要有感染、化学、肿瘤、内分泌等。该病发病率低,临床表现复杂多样,目前认为有内脏器官受累者预后偏差。SSc早期即可累及多个器官系统,临床病程多变[4]。其中呼吸系统较易受累,主要表现为肺间质纤维化和肺动脉高压。本例患者合并肺间质病变,院外肺功能提示弥散功能轻度减低,肺通气功能正常,此次病程中无呼吸困难等临床表现。其次SSc患者的消化系统表现多样,包括胃食管反流、胃窦血管扩张症、腹痛、腹泻、便秘等,本例患者既往长期有腹胀、消化不良、腹泻等病史,既往上消化道造影检查提示食管蠕动减弱。心脏受累可表现为心包积液、心脏传导系统异常,本例患者既往超声提示心包积液,心电图提示完全性右束支传导阻滞。吉兰-巴雷综合征(Guillain-Barre syndrome,GBS)病理改变是周围神经组织中小血管周围淋巴细胞与巨噬细胞浸润以及神经纤维的脱髓鞘,严重时可出现继发性轴索变性。GBS的诱因主要是呼吸道感染和(或)胃肠道感染,而消化道空肠弯曲菌感染很有可能发展为轴索型,预后较差[5]。本例患者神经苷脂抗体检测均为阴性,而Yamaguchi等[1]报道的病例发病前有带状疱疹病毒感染,神经苷脂抗体抗GM1、GD1b IgM抗体阳性,另外1例患者未报告相关抗体。
据报道1%~40%的SSc患者可发生神经系统并发症[6],而具有抗U1RNP抗体和抗Scl70抗体的患者发生神经系统并发症的风险更高。SSc中枢神经系统累及多表现为焦虑、抑郁等精神症状,也可表现为头痛、认知障碍、癫痫发作、器质性脑综合征等,神经影像学检查可见脑白质高信号、脑血管病变、脑萎缩等,脑单光子发射计算机断层显像可见局灶性或弥漫性灌注不良。SSc周围神经系统病变常见的有三叉神经病变、周围感觉运动障碍、卡压性神经病变等,也可累及自主神经系统导致血压变异性升高,副交感神经功能紊乱等。血管病变常加重SSc神经系统病损程度。其可能的病理机制是:内皮细胞损伤导致的血管通透性增加与单核细胞浸润相关,进而导致血管周围炎性细胞浸润、血管内膜增厚和血管狭窄,久之,小动脉失去弹性、血管内膜中膜变得疏松,导致其更容易发生闭塞,促血管生成因子和抗血管生成因子的过度表达,进一步加重血管生成失调,发生缺氧状态,形成无血管区,导致相应神经营养障碍。自身抗体在SSc病变中也发挥重要作用[7]。约95%的SSc患者可检测到自身抗体,在大多数情况下,自身抗体具有高度特异性。在SSc患者血清中可检测到抗成纤维细胞抗体、抗血管紧张素抗体、抗内皮素抗体,其被认为与疾病严重程度相关,进一步研究发现,抗血小板衍生生长因子受体的抗体是SSc的特异性标志,其可产生活性氧,刺激肌成纤维细胞分化和1型胶原基因表达[8]。
目前关于SSc合并GBS的机制尚不清楚,但机体免疫系统产生的抗体在二者中发挥重要作用。其可能的机制为:(1)SSc相关抗体与体内具有相同抗原决定簇的自身组织发生免疫反应,从而导致神经系统免疫损伤。前期感染病史如上呼吸、胃肠道感染是潜在诱因。本例患者SSc继发GBS,分型为急性炎症性脱髓鞘性多发性神经病,该患者既往长期服用免疫抑制剂及激素治疗SSc,导致自身免疫功能紊乱,也可能是发生GBS的重要原因。(2)本例患者神经节苷脂抗体检测均为阴性,但抗核抗体、Ro-52、AMA M2、PM-Scl检测阳性,且神经系统运动功能恢复迟缓,结合既往报道,当SSc与GBS并存时,后者可能为系统性硬化症周围神经受累表现,但仍需进一步临床研究证实。回顾文献发现只有1例患者完善了腓肠神经活检,发现节段性髓鞘再形成,这表明存在亚临床神经病变,推测其与SSc相关,提示既存神经病变很可能加重了GBS全身症状的严重程度,影响预后。
4 结 论
SSc合并GBS是临床很罕见的一类疾病,其具体的发生机制目前尚不清楚,自身免疫反应及相关细胞因子在这类疾病发生发展中发挥重要作用。GBS可能是SSc的周围神经病变,但仍需与其他周围神经病鉴别,后期研究需进一步明确二者之间的发病机制。其次系统性硬化症患者诊治过程中需加强神经系统损害的筛查,早期识别、减少误诊、积极治疗、减少复发是改善本类疾病患者预后和提高生活质量的重点。
猜你喜欢
中国临床医学影像杂志(2019年5期)2019-08-27 02:47:44
中国神经精神疾病杂志(2018年11期)2018-12-11 05:30:56
高中生·天天向上(2017年4期)2017-06-09 02:37:17
武警医学(2016年3期)2016-03-13 11:14:51
中国实用神经疾病杂志(2016年5期)2016-01-26 15:08:10
电影故事(2015年43期)2015-11-16 05:51:18
西安交通大学学报(医学版)(2015年2期)2015-02-28 17:59:20
西南军医(2014年5期)2014-04-25 07:42:33
卒中与神经疾病(2014年5期)2014-03-23 13:17:02
