
可交联型两性双亲分子的合成及其溶致液晶相结构的研究
2021-08-30 04:47吴晗宇冯训达
液晶与显示 2021年8期
吴晗宇, 冯训达*
(1. 东华大学 材料科学与工程学院 纤维材料改性国家重点实验室,上海 201620;2. 东华大学 先进低维材料中心,上海 201620)
1 引 言
提高材料内部结构的有序性是一种常见的提升材料性能的方法。与无机纳米材料相比,有机聚合物纳米材料在获取高度有序结构上难度较大。如何在聚合物中构筑有序的纳米结构,实现对聚合物纳米材料的结构操纵、性质调控及功能的多样化,已成为化学、材料科学及生物医药等多学科交叉领域的研究热点[1-2]。
利用溶致液晶相的原位聚合固定化来制备具有有序纳米结构的聚合物材料是一种行之有效的方法[3-4]。简单来说,首先设计和合成具有可聚合基团的小分子液晶基元,例如热致液晶分子或者是形成溶致液晶所需的表面活性分子等;这些液晶基元进一步自组装成高度有序的液晶相,最后通过光引发自由基聚合反应,有序结构得到保存,从而在聚合物中构筑有序纳米结构。目前通过这一策略,已经制备了具有一维(六方柱状结构)、二维(层状结构)以及三维(双连续立方结构)有序纳米结构的聚合物薄膜材料,并应用于膜分离和离子传导[5-10]。由于聚合反应涉及化学键的断裂与生成,在聚合过程中分子的构象会发生变化,分子的聚集状态很容易发生改变,导致聚合后溶致液晶有序纳米结构不复存在[11]。
为了避免聚合反应前后溶致液晶相态的破坏,可以在溶致液晶体系中引入交联剂[10],由于外加交联剂会破坏溶致液晶相态,在体系中引入的交联剂含量不能过多,因此外加交联剂的方法往往也无法实现溶致液晶相态的聚合固定化。对于可聚合型溶致液晶,在体系中加入的光引发剂含量极少,其质量占溶致液晶总质量的0.1%左右,一般不会对溶致液晶的相态产生影响。Gin等人合成了可聚合型多碳链的两亲分子,在两条、或者三条碳链的末端接上可聚合基团,因此配备出的液晶结构具有优良的结构固化能力。然而,此类可交联的分子结构往往使得分子的疏水体积比例增大,趋于形成疏水朝外、亲水朝内的反相液晶结构[12]。
为此,我们设计合成了一种新型的具有双交联官能团的两性双亲离子溶致液晶基元。在单一碳链末端引入两个可聚合的甲基丙烯酸酯基团,此分子结构不仅保持了单链双亲分子的几何结构,同时在聚合反应时能迅速将溶致液晶结构固化保留下来,制备具有有序结构的聚合物材料。该分子独特的分子结构为溶致液晶相态的光固化提供了一条新的分子设计思路。
2 实 验
2.1 实验原料
1,10-二溴癸烷(>95%,国药集团);丙二酸二乙酯(97%,阿拉丁);乙醇钠(96%,阿拉丁);硼氢化钠(98%,阿拉丁);二甲胺水溶液(质量分数为40%,阿拉丁);甲基丙烯酰氯(95%,阿拉丁);三乙胺(≥98%,国药集团);1,3-丙烷磺酸内酯(99%,阿拉丁);甲醇(≥99.5%, Greagent);乙醇(≥99.5%, Greagent);乙腈(≥98.5%,国药集团)通过活化4A分子筛干燥回流重蒸后使用。
2.2 测试仪器
1H NMR核磁共振氢谱:布鲁克(Bruker) AVANCE NEO 400 MHz,测试时使用氘代氯仿(CDCl3,溶剂峰7.26)或氘代水(D2O,溶剂峰4.80)作为测试溶剂。Olympus BX530型偏光显微镜(POM),用于观察液晶分子的溶致液晶织构。
2.3 可聚合型两性离子液晶基元DMAC11-APS的合成
可聚合型两性离子液晶基元DMAC11-APS的合成反应式如图1(b)所示,其中,DMA表示分子烷基链末端的两个甲基丙烯酸酯基团,C11表示链长为11的烷基链,APS表示分子中磺基甜菜碱型的离子部分。液晶基元DMAC11-APS的具体合成步骤及各中间体的分子结构表征如下。
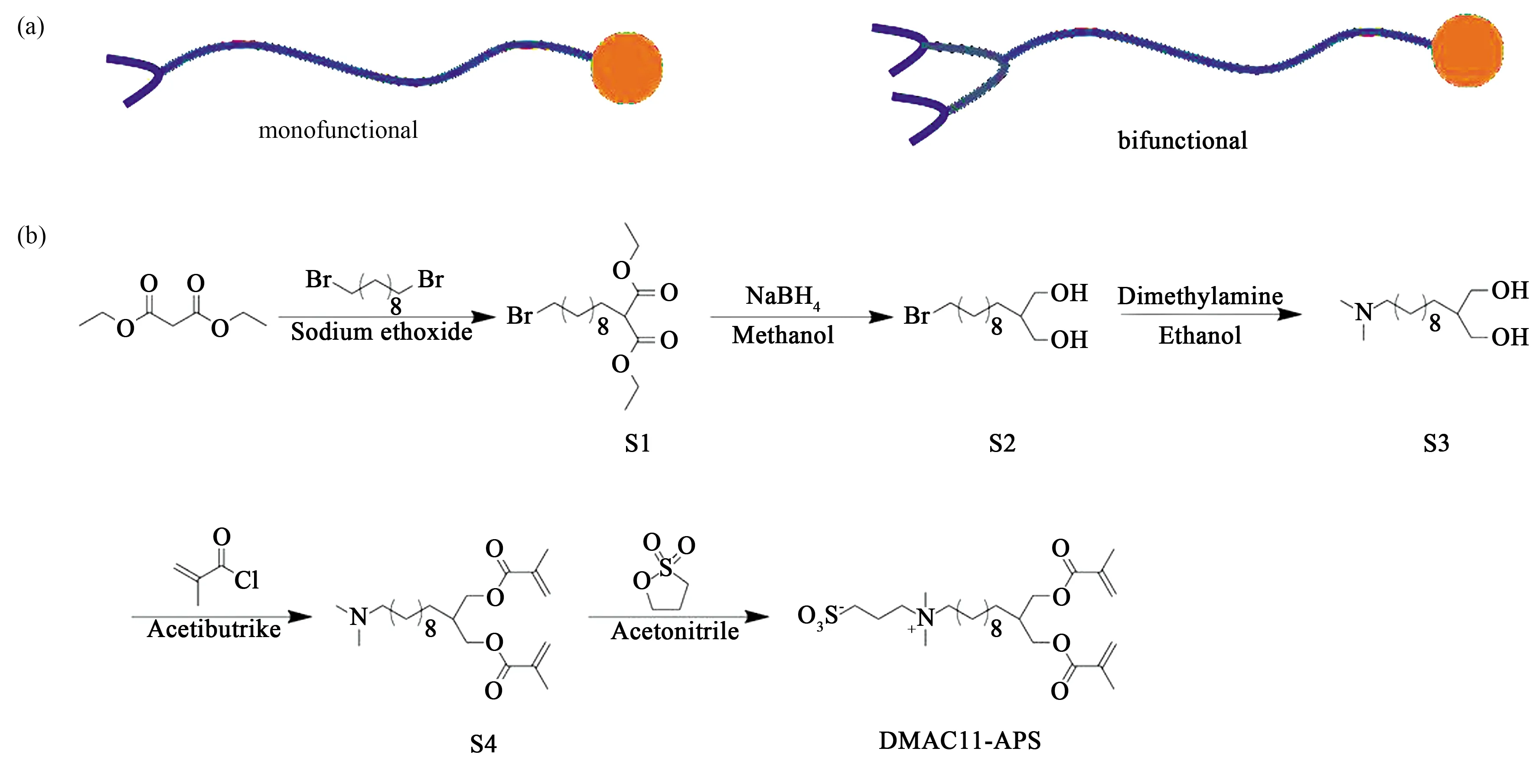
图1 (a)传统的具有单可聚合官能团的液晶基元及本文设计与合成的末端具有双可聚合官能团的结构示意图;(b)可聚合型两性离子液晶基元DMAC11-APS的合成路线。Fig.1 (a)Zwitterionic mesogens with monofunctional and bifunctional polymerizable groups; (b) Synthesis of polymerizable zwitterionic mesogen with double methacrylate groups.
2.3.1 中间体S1的合成
乙醇钠(15.0 g,0.22 mol)溶于100 mL乙醇,然后向溶液中滴加丙二酸二乙酯(48.0 g, 0.3 mol)。 然后将混合物在室温搅拌10 min。滴加1,10-二溴代癸烷(59.6 g, 0.2 mol),并在90 ℃下回流24 h。减压蒸出溶剂,将剩余物溶于300 mL HCl(1 mol/L)水溶液中。用乙醚萃取,并用无水硫酸钠干燥。将未反应的丙二酸二乙酯在120 ℃下减压蒸馏除去。粗产物通过硅胶柱色谱法纯化,用己烷作为洗脱液分离出1,10-二溴代癸烷,然后使用乙酸乙酯∶己烷(1∶30)洗脱,得到中间体S1,为无色油状液体(30.49 g, 40%)。1H NMR (400 MHz, Chloroform-d)δ∶4.13 (qd,J= 7.1, 1.3 Hz, 4H), 3.34 (t,J= 6.9 Hz, 2H), 3.24 (t,J= 7.6 Hz, 1H), 1.87 ~1.73 (m, 4H), 1.39~ 1.32 (m, 2H), 1.30 ~1.14 (m, 18H)。
2.3.2 中间体S2的合成
中间体S1(10.0 g,26.36 mmol)溶解在250 mL甲醇中,在剧烈搅拌的同时将NaBH4(9.97 g,263.62 mml)缓慢且小心地添加至甲醇溶液中。在65 ℃下回流反应1 h。然后将80 mL水加入到反应瓶中,并在室温下再搅拌1 h。滤出无机盐,产物用乙醚萃取并用无水硫酸钠干燥。粗产物通过柱色谱法纯化,使用二氯甲烷∶乙酸乙酯(5∶1)作为洗脱剂,得3.5 g白色固体S1,收率为45%。1H NMR (400 MHz,Chloroform-d)δ:3.81 (dd,J= 10.6, 3.7 Hz, 2H), 3.65 (dd,J= 10.6, 7.6 Hz, 2H), 3.40 (t,J= 6.9 Hz, 2H), 2.47 (s, 2H), 1.84 (dt,J= 14.5, 7.0 Hz, 2H), 1.80 ~ 1.71 (m, 1H), 1.41 (dd,J= 9.7, 5.3 Hz, 2H), 1.26 (m,J= 11.5, 8.8 Hz, 14H)。
2.3.3 中间体S3的合成
中间体S2(2.0 g,6.77 mmol)和20 mL、质量分数为40%的二甲胺水溶液溶解于100 mL乙醇中,80 ℃回流反应24 h。反应完全后,减压蒸馏除去溶剂,将残余物溶于100 mL,质量分数为5% NaHCO3水溶液,氯仿萃取产物,有机相用无水硫酸钠干燥并浓缩,得1.55 g中间体S3,为白色固体,收率为88%。1H NMR (400 MHz, Chloroform-d)δ:3.80 (dd,J= 10.6, 3.7 Hz, 2H), 3.63 (dd,J= 10.6, 7.7 Hz, 2H), 3.49~3.19 (broad, 2H), 2.34 ~2.27 (m, 2H), 2.25 (s, 6H), 1.76 (ddt,J= 10.8, 7.2, 3.6 Hz, 1H), 1.53 ~ 1.43 (m, 2H), 1.37 ~1.16 (m, 14H)。
2.3.4 中间体S4的合成
中间体S3(1.4 g, 5.4 mmol)和三乙胺(1.64 g,16.19 mmol)溶解在100 mL乙腈中,甲基丙烯酰氯(1.35 g, 12.95 mmol,溶解于10 mL乙腈)在冰浴条件下缓慢滴加到反应瓶中。滴加完成后,室温反应12 h。反应结束后,将反应液倒入100 mL、质量分数为5%的NaHCO3水溶液中,用二氯甲烷萃取3次,并用无水硫酸钠干燥。柱色谱法纯化粗产物,洗脱剂为二氯甲烷,得2.0 g浅红色液体S4,收率为94%。1H NMR (400 MHz, Chloroform-d)δ:6.10 (s, 2H), 5.56 (s, 2H), 4.21~ 4.08 (m, 4H), 2.84~ 2.79 (m, 2H), 2.66 (s, 6H), 2.10 (p,J= 6.2 Hz, 1H), 1.94 (s, 6H), 1.73 (q,J= 5.2, 3.5 Hz, 2H), 1.28 (m,J= 21.5 Hz, 16H)。
2.3.5 可交联型两性双亲分子DMAC11-APS的合成
中间体S4(2.0 g,5.06 mmol)、1,3-丙烷磺内酯(1.24 g,10.11 mmol)、4-甲氧基苯酚(10 mg,作为阻聚剂)溶解在100 mL乙腈中,并在氩气保护下40 ℃反应4天。反应结束后,将反应液滴加到大量冷乙醚中以沉淀产物。离心干燥得1.6 g淡黄色蜡状固体DMAC11-APS,收率为57%。1H NMR (400 MHz, Chloroform-d)δ:6.10 (s, 2H), 5.56 (s, 2H), 4.21 ~ 4.08 (m, 4H), 3.57~ 3.45 (m, 2H), 3.41~3.27 (m, 2H), 3.14 (s, 6H), 2.98 (t,J= 7.0 Hz, 2H), 2.32~2.16 (m, 2H), 1.93 (s, 6H), 1.78 (s, 2H), 1.72 ~1.59 (m, 2H), 1.34 (m,J= 27.4 Hz, 14H)。
可交联型两性双亲分子DMAC11-APS和中间体S4的核磁共振氢谱(1H NMR)如图2所示,从图中可以看出,季胺化反应发生后,与氮原子相连的两个甲基(-CH3)的化学位移向低场移动,这证实了可交联型两性双亲分子DMAC11-APS的成功合成。
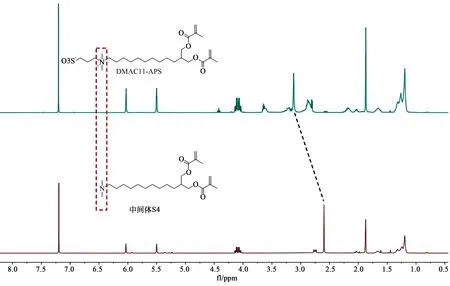
图2 可交联型两性双亲分子DMAC11-APS和中间体S4的核磁共振氢谱图Fig.2 1H NMR spectra of DMAC11-APS and intermediate S4
3 结果与讨论
3.1 液晶样品的制备及相图的绘制
称取一定量的液晶基元DMAC11-APS放于2 mL离心管中,按照一定的质量比,用移液枪移取对应质量的去离子水溶液至离心管中,然后将离心管中的样品离心、搅拌、加热,重复数次,直至样品完全均匀。以3%(质量分数)为间隔,配置不同质量分数的溶致液晶样品,通过样品在偏光显微镜中的织构判断其溶致液晶相态。六方柱状相(Hexagonal)为扇状织构;双连续立方相(Bicontinuous cubic)为无偏光,且粘度很高;层状相为油丝状织构或马耳他十字织构。
可聚合型两性离子液晶基元DMAC11-APS与水在室温下的相图如图3 (a)所示。我们发现,双亲分子交联剂DMAC11-APS与水共混时,当浓度区间位于质量分数0~63%时,共混物为可流动的、粘度低的液体。同时,在偏振光显微镜下,样品无双折射性。因此,我们判定此浓度区间为无序的胶束相(Iso)。在浓度区间为质量分数63%~74.5%,共混物的粘度有所上升,偏振光显微镜中部分区域出现了双折射的织构。因此,该浓度区间为无序相与液晶相的共存区。
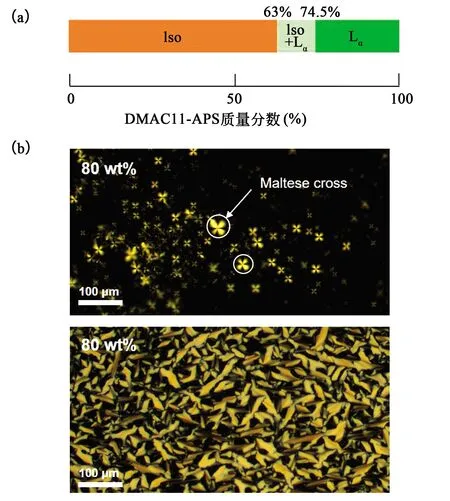
图3 (a)可聚合型两性离子液晶基元DMAC11-APS与水在室温下的相图,Iso表示无序胶束相,Lα表示层状相;(b)液晶基元DMAC11-APS与水形成层状相的偏光显微镜织构,图中可见明显的马耳他十字织构和破碎焦锥织构。Fig.3 (a) Binary phase diagram for mixtures of DMAC11-APS and water at room temperature. Iso: disorder micellar phase; Lα: lamellar phase; (b) POM images of Maltese cross texture and broken focal conic texture confirm the formation of lamellar phase (Lα).
而当浓度区间大于质量分数74.5%时,共混物表现为较为粘稠。偏振光显微镜的图像表明,液晶呈现典型的层状液晶(Lα)织构,表现为马耳他十字(Maltese cross)、破碎焦锥(Broken focal conic)[13]。
3.2 液晶基元DMAC11-APS的溶致液晶相行为分析
可聚合型两性离子液晶基元DMAC11-APS与水仅仅只形成层状相(Lα),不会形成六方柱状相(H1)。溶致液晶分子的相行为可以根据临界堆积参数理论(Critical Packing Parameter, CPP)进行粗略的判断,临界堆积参数CPP本质上是对分子亲水和疏水部分的简单几何形态描述[14]。当0< CPP≤ 1/3时,分子在水溶液中只能形成球状胶束;当1/3< CPP≤ 1/2时,分子趋向于形成球状胶束和六方柱状相;当1/2< CPP≤1时,分子易于形成层状相。根据临界堆积参数理论(Critical Packing Parameter, CPP),我们可以推测可聚合型两性离子液晶基元DMAC11-APS的临界堆积参数CPP介于1/2和1之间。液晶基元DMAC11-APS的结构示意图如图4 (a)所示,液晶基元DMAC11-APS在水中形成层状相(Lα)的分子排列如图4 (b)所示,分子排列为双分子层,亲水的头部排列在外部与水作用,层与层之间为水分子。
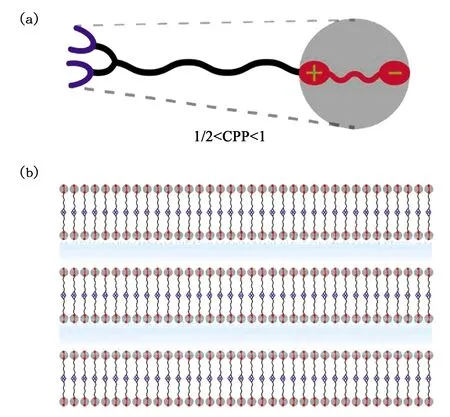
图4 (a) 液晶基元DMAC11-APS的结构示意图;(b)液晶基元DMAC11-APS在水中形成层状相(Lα)的分子排列示意图。Fig.4 (a) Schematic illustration of the molecular structure of DMAC11-APS; (b) Schematic illustration of the mesogen DMAC11-APS assembled in a lamellar phase(Lα) by hydration.
4 结 论
设计并合成了一种可聚合型两性离子液晶基元,并进一步探究了该液晶基元的溶致液晶相态。液晶基元DMAC11-APS与水在室温下仅形成层状相液晶,根据临界堆积参数理论,分析推测液晶基元DMAC11-APS的临界堆积参数CPP值介于1/2与1之间。液晶基元DMAC11-APS的独特分子结构为溶致液晶相态的光固化提供了一条新的分子设计思路,可实现溶致液晶有序结构的高保真光固化,从而制备具有有序纳米结构的聚合物材料。鉴于该液晶基元与水自组装形成的溶致液晶仅存在层状相(Lα),可将其当作交联剂加入到其他可聚合型溶致液晶体系中,实现对其他溶致液晶相态的光固化,制备具有其他有序纳米结构的聚合物材料。
猜你喜欢
计算机工程与应用(2023年1期)2023-01-13
宝钢技术(2022年3期)2022-07-12
科学导报(2018年30期)2018-05-14
材料科学与工程学报(2016年4期)2017-01-15
光学精密工程(2016年4期)2016-11-07
高原山地气象研究(2016年3期)2016-02-28
船海工程(2015年4期)2016-01-05
中国塑料(2015年4期)2015-10-14
石油化工应用(2014年11期)2014-03-11
