
草原1号杂花苜蓿花蕾转录组SSR序列特征
2021-08-26 07:06石凤翎乌日娜
西北农林科技大学学报(自然科学版) 2021年8期
徐 舶,石凤翎,乌日娜,乔 雨,王 佳
(内蒙古农业大学 草原与资源环境学院,草地资源教育部重点实验室,内蒙古 呼和浩特 010019)
苜蓿(Medicago)为豆科多年生草本植物, 是世界上栽培历史最悠久、营养价值最高的优良牧草之一, 具有生态和商业双重价值[1]。栽培苜蓿的主要育种目标是提高饲草品质和增加产量[2],但由于苜蓿基因组高度杂合、遗传分析复杂,导致品种更新换代慢,很难提高草料品质和产量[3]。此外,苜蓿的品质和产量性状受数量性状基因座(QTL)的调节,而QTL会受到环境变化的影响[4],这使得对品质和产量性状的选择育种变得更加困难。分子标记与植物基因型密切相关,受环境因素的影响较小[5],目前已被广泛应用于辅助育种[6]、品种鉴定[7]、基因组图谱构建[8]及靶区域的位置克隆[9]等研究,这为栽培苜蓿的改良提供了一条新的途径[10]。
简单重复序列(SSR)或微卫星SSR是DNA序列中1~6个核苷酸的短串联重复序列,其重复数目高度可变,而其侧翼区域高度保守[11]。SSR标记是基于聚合酶链反应(PCR)的高度可复制的共显性标记,因其丰富的多态性和多等位性而被广泛用于种质遗传多样性分析和标记辅助选择育种[12],利用高通量测序技术开发SSR引物在褐沙蒿[13]、红豆草[14]、橡胶草[15]等牧草中均有报道。当前,在苜蓿中应用的SSR标记多从紫花苜蓿[16]和蒺藜苜蓿[17]基因组文库中获得,其标记数量较为有限,且随着近几年我国苜蓿品种登记数量的逐渐增多(目前我国登记品种超过100个),为满足苜蓿种质资源鉴定及提高育种选择效率的需要,有必要开发新的分子标记。
本研究基于草原1号杂花苜蓿(MedicagovariaMartin.‘Caoyuan No.1’)花蕾的转录组高通量测序结果,利用 MicroSAtel- lite(MISA)软件搜索草原1号杂花苜蓿 SSR 位点信息,并对SSR分布、组成及其序列特征进行分析,以期为栽培苜蓿遗传多样性研究、功能基因的定位和克隆、分子辅助育种等提供更多实用的分子标记。
1 材料与方法
1.1 试验材料
根据前人的研究结果[18],选择草原1号杂花苜蓿花粉母细胞形成时期(花蕾长0.8~2 mm)、单核花粉粒时期(花蕾长2~3 mm)和花粉成熟期(花蕾长4~5 mm)3个发育时期的花蕾。将同一株同一时期的花蕾剥下,去除花萼,混合后放入液氮中速冻,于超低温(-80 ℃)冰箱中保存,用于后续转录组的测序。
1.2 试验方法
1.2.1 转录组文库构建与测序 利用天根提取试剂盒分别对各单株各时期的材料进行总RNA提取,再采用TruSeqTM RNA sample preparation Kit(Illumina,San Diego,CA)试剂盒进行RNA文库的建立。首先利用带有Oligo(dT)的磁珠从5 μg总RNA中富集有poly-A尾巴的Mrna,之后加入fragmentation buffer,将mRNA随机断裂成200 bp左右的小片段,接着采用 SuperScript double-stranded cDNA synthesis kit (Invitrogen,CA)试剂盒,加入六碱基随机引物(Illumina),以小片段的mRNA为模板反转合成一链cDNA,随后进行二链合成,形成稳定的具有双链结构的cDNA。在双链cDNA中加入End Repair Mix进行末端补平,并在3′端加上一个A碱基,用于连接Y字形的接头。cDNA 经过PCR 富集后,利用2%琼脂糖凝胶回收长度200~300 bp的目的条带。经TBS380(Picogreen)定量后,将文库用上海美吉生物公司的Illumina HiSeq X Ten测序平台进行高通量测序,测序读长为PE 150。
1.2.2 质量评估与拼接 完成各样品的高通量测序后,对产生的大量原始读序(Raw reads)进行过滤筛选。去除含接头序列、未插入序列以及含碱基信息比例超过10%和长度低于20 bp的序列,由此获得高质量的过滤基因片段(clean reads)。将过滤所得的基因片段通过短序列组装软件Trinity进行从头拼接组装(de novo assembly),取每条基因中最长的转录本(Transcripts)作为单基因簇(Unigene)[19],以拼接成的非冗余草原1号杂花苜蓿花蕾Unigene作为后续分析的参考序列。
1.2.3 SSR的筛选和统计分析 使用软件MISA对组装的非冗余 Unigene进行搜索,寻找Unigene中的 SSR位点。检索标准:精确型(perfect)单、二、三、四、五、六核苷酸基序(motif)至少分别重复10,6,5,5,5和5次;复合型(compound)核苷酸2个位点间不超过100 bp[20]。用Microsoft Excel 2010对数据进行整理和绘图。
2 结果与分析
2.1 草原1号杂花苜蓿转录组SSR分析
由表1可知,对草原1号杂花苜蓿进行高通量测序后,共得到286 409条非冗余Unigene,总长度为335 218 367 bp,平均长度为1 170 bp。按照搜索标准对非冗余序列进行筛选,共搜索到57 368个SSR位点,分布在46 337条Unigenes上。SSR位点的出现频率和发生频率分别为20.03%和16.18%。此外,含复合型SSR位点的Unigene序列数有3 519条,含1个以上SSR位点的Unigene序列有6 117条,占Unigenes 序列总数的2.14%。

表1 草原1号杂花苜蓿转录组中SSR搜索分析结果Table 1 Search and analysis of SSR in transcriptome of M.varia Martin.‘Caoyuan No.1’
2.2 草原1号杂花苜蓿转录组SSR重复类型分析
表2显示,草原1号杂花苜蓿转录组SSR位点类型丰富,且各重复类型的出现频率具有明显差异。所有SSR重复类型中,所占比例和出现频率最高的是单核苷酸重复,分别为68.45%和13.71%;其次为三核苷酸和二核苷酸重复,所占比例分别为16.47%和13.28%;四、五、六核苷酸的数量相对较少,所占比例和出现频率总计为1.80%和0.36%;单核苷酸重复的SSR数量约是三核苷酸重复的4倍,约是六核苷酸重复的577倍。从总体分布情况来看,每8.54 kb出现1个单核苷酸SSR位点,每4 929.68 kb出现1个六核苷酸位点,平均每5.84 kb就出现1个SSR位点。
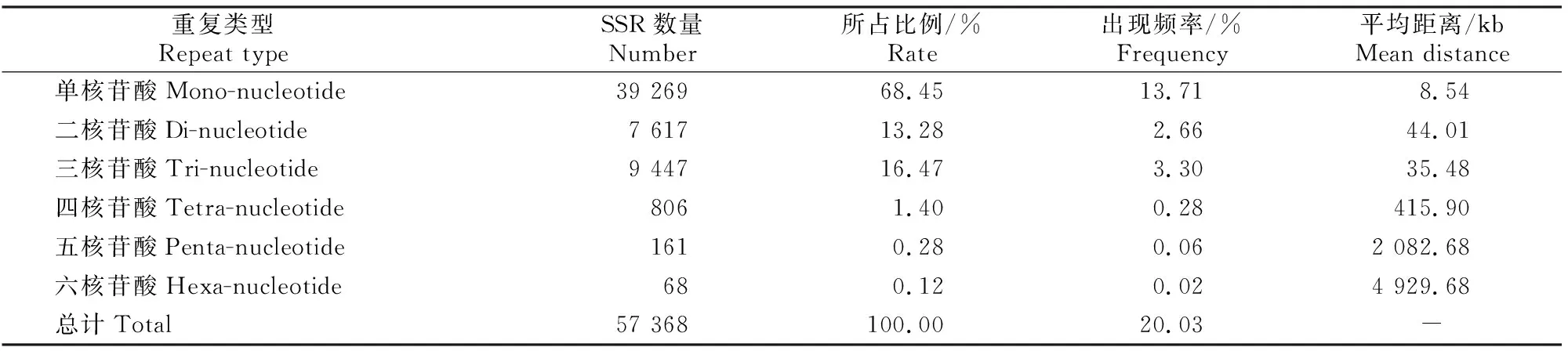
表2 草原1号杂花苜蓿转录组SSR重复类型的数量和分布特点Table 2 Number and distribution characteristics of SSR repeat types in transcriptome of M.varia Martin.‘Caoyuan No.1’
2.3 草原1号杂花苜蓿转录组SSR重复基元类型和比例
表3显示,在草原1号杂花苜蓿转录组中检测到由129种重复基元构成的57 368条SSR,其中单、二、三、四、五、六核苷酸重复基元类型数分别为2,4,10,28,45和40,其分布总频率为20.03%。在核苷酸重复基元类型中,单核苷酸重复基元A/T数量最多(39 099个),占比高达68.15%;其次为二核苷酸中的AG/CT基元(数量3 766,占比6.56%)和三核苷酸中的 AAC/GTT基元(数量2 319,占比4.04%)。其中,二核苷酸中的优势基元为AG/CT(占总SSR位点数的6.56%,下同)、AC/GT(3.68%)、AT/AT(2.96%); 三核苷酸中的优势基元为AAC/GTT(4.04%)、AAG/CTT(3.45%)、ATC/ATG(2.28%)、AAT/ATT(1.98%); 四核苷酸中的优势基元为AAAT/ATTT(0.39%);五、六核苷酸数量较少,其各类型重复基元所占比例均低于1%。
表3还显示,6种重复类型核苷酸的分布频率为0.01%~13.65%,其中单核苷酸重复基元A/T的分布频率最高(13.65%),二核苷酸重复基元AG/CT(1.31%)、三核苷酸重复基元AAC/GTT(0.81%)次之,四、五、六核苷酸分布频率均较低,仅为0.01%~0.16%。总体从基元类型和所占比例来看,草原1号杂花苜蓿转录组中SSR的五核苷酸重复基元种类最多,而单核苷酸重复基元数量最多。

表3 草原1号杂花苜蓿转录组中SSR的重复基元类型及数量Table 3 Type and number of SSRs repeat motifs of M.varia Martin.‘Caoyuan No.1’
2.4 草原1号杂花苜蓿转录组SSR重复次数及分布
从草原1号杂花苜蓿SSR位点重复次数的分布结果(图1)可以看出,转录组基元数量和比例随着重复次数的增加总体呈减少趋势,SSR重复次数主要集中在6~15次(共44 422条),占总重复次数的77.44%;低重复次数(1~5次)的SSR位点为5 664个(9.87%); 较高重复次数(重复次数>15次)的SSR位点有7 282个,占总SSR位点数的12.69%。

图1 草原1号杂花苜蓿转录组SSR重复次数的分布Fig.1 Distribution of SSR repeat types in transcriptome of M.varia Martin.‘Caoyuan No.1’
由表4可知,6种核苷酸重复类型的重复次数与整体重复次数表现出相同的变化趋势,均随着重复次数的增加而逐渐降低,以较低(1~10次)或一般(11~15次)重复次数为主。其中单核苷酸和二核苷酸基元重复次数主要集中在6~15次,主要重复基元比例分别为83.61% 和89.79%;三核苷酸基元重复次数主要集中在1~10次,主要重复基元比例为97.40% ,单、二、三核苷酸主要重复基元总计占总SSR位点数的85.19%;四、五、六3种核苷酸的基元重复次数主要集中在低重复水平(1~10次),其主要重复基元数量分别为797,160和66个,占总SSR的比例分别为1.39%,0.28%和0.12%。
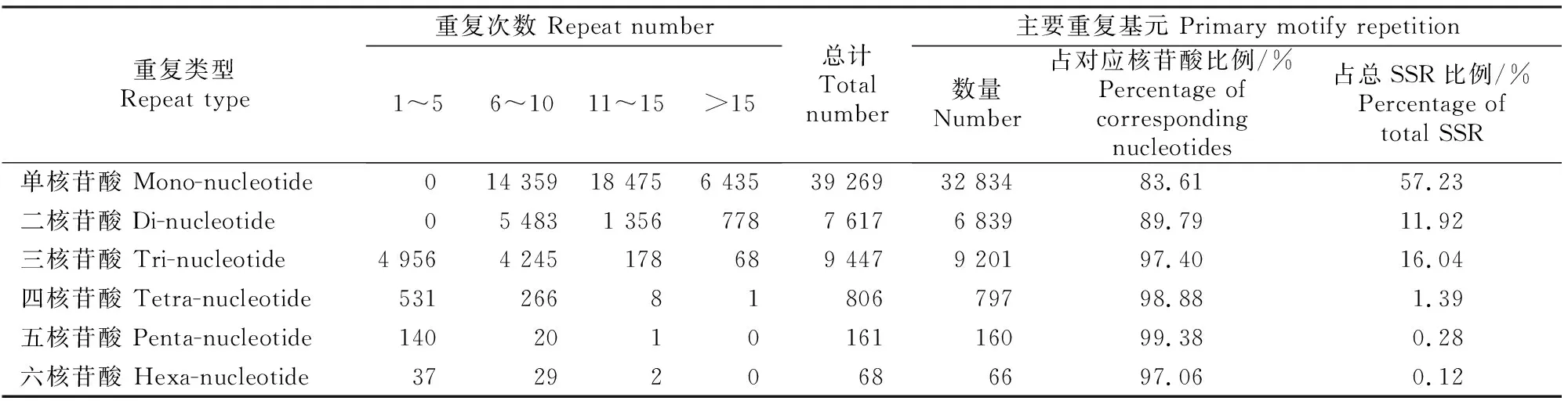
表4 草原1号杂花苜蓿转录组SSR不同基元的重复次数Table 4 Repeat times of different SSR motifs of M.varia Martin.‘Caoyuan No.1’
2.5 草原1号杂花苜蓿转录组SSR长度的分布
SSR基元长度是影响其多态性的主要因素之一。过滤掉长度<12 bp的片段后,对草原1号杂花苜蓿转录组不同重复类型SSR的长度分布进行分析,结果见表5。

表5 草原1号杂花苜蓿转录组不同重复类型SSR的长度分布Table 5 Length distribution of SSR different repeat types in transcriptome of M.varia Martin.‘Caoyuan No.1’
由表5可知,草原1号杂花苜蓿SSR长度为12~365 bp。二核苷酸SSR长度为12~80 bp,平均长度为45 bp,主要集中在12~24 bp,总计占二核苷酸总数的85.04%; 三核苷酸重复中SSR长度为15~93 bp,平均长度53 bp,以15 bp的核苷酸最多(4 219个),占三核苷酸总数的52.13%; 四、五、六3种核苷酸SSR长度分别为20~72,25~60,30~60 bp,平均长度分别为40,39和45 bp,其占比最大的SSR长度分别为20 bp(65.19%),25 bp(86.33%)和30 bp(55.00%)。检测到的复合型核苷酸长度主要集中在20~120 bp,总计占复合型核苷酸总数的92.92%。
过滤掉长度<12 bp的片段后,草原1号杂花苜蓿转录组中SSR基序长度分布如图2所示。图2显示,除去长度<12 bp的片段后,草原1号杂花苜蓿转录组的SSR长度主要集中在12~40 bp,占总SSR位点数的55.58%(31 883条),其中以长度15 bp的SSR数量最多,共4 219条,占比7.35%,均为三核苷酸重复。长度为12~20 bp的SSR有25 820条,占总SSR位点数的44.99%;长度21~40 bp的SSR有6 063条,占总SSR位点数的10.57%;长度在41~60 bp的SSR有1 212条,占总SSR位点数的2.11%;长度在160 bp以上的SSR有48条,占总SSR位点数的0.08%。经统计,长度在20 bp以上的SSR共有9 166条,占总SSR位点数的15.98%,这部分序列可能具有较高的多态性潜能。
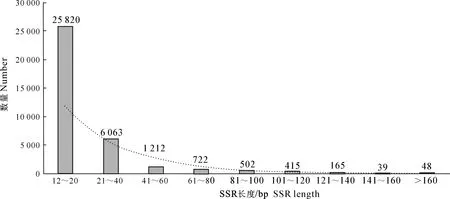
图2 草原1号杂花苜蓿转录组中 SSR 基序长度的分布Fig.2 SSR motif length distribution in transcriptome of M.varia Martin.‘Caoyuan No.1’
3 讨论与结论
基于转录组测序结果的分子标记技术,已经广泛应用于物种基因探索与功能注释、遗传多样性分析以及辅助选择育种研究[21]。本研究通过对草原1号杂花苜蓿花蕾的高通量测序,共计得到非冗余序列Unigene 286 409条,总长度为335 218 367 bp。从所有非冗余Unigene中共搜索到SSR位点57 368个,SSR出现频率为20.03%,高于马蓝(16.49%)[21]、蒙农红豆草(15.42%)[14]、褐沙蒿(13.02%)[13]和鱼腥草(7.51%)[22],低于夏枯草(27.5%)[23]、蒙古黄芪(31.26%)[24]和橡胶草(35.2%)[15]。草原1号杂花苜蓿SSR平均分布距离为5.84 kb,高于橡胶草(1.86 kb)[15]、白沙蒿(4.38 kb)和乌丹蒿(4.37 kb)[25],低于夏枯草(5.93 kb)[23]、蒙古黄芪(6.51 kb)[24]和马蓝(7.48 kb)[21]。由此可见,SSR的分布具有物种特异性,且各研究报道的结果也与测序文库的大小和SSR搜索标准不同等因素有关。此外,草原1号杂花苜蓿与同为豆科属植物的蒙农红豆草的分布距离(5.86 kb)极为相近,两者可进行相关的分子标记开发以及类比分析。
研究发现,大多数植物的SSR以单核苷酸、二核苷酸和三核苷酸重复基元为主,但是主导重复基元的类型因物种差异、测序文库数据量多少和搜索标准而有所不同[21]。本研究中,草原1号杂花苜蓿花蕾转录组SSR的重复基元类型主要是单核苷酸(68.45%),其次为三核苷酸(16.46%)和二核苷酸(13.27%),这与紫花苜蓿(单核苷酸占比48.21%)[26]、橡胶草(单核苷酸占比77.47%)[15]的研究结果一致。而马蓝[21]、蒙药冷蒿[27]以二核苷酸和三核苷酸为主,蒙农红豆草[14]、褐沙蒿[13]以三核苷酸为主,寒兰[28]、夏枯草[23]以二核苷酸为主,这种优势重复基元的不同也体现了物种的特异性。在本研究搜索到的SSR位点中,单核苷酸和二核苷酸重复的优势基元分别为A/T和AG/CT,与前人报道[26]一致,但三核苷酸重复的优势基元为AAC/GTT,有别于常见的其他豆科物种(紫花苜蓿[26]、蒙农红豆草[14]优势基元均为AAG/CTT),利用这一特点可以开发出更丰富、更实用的SSR新位点。
SSR位点的多态性主要由基元重复次数和长度大小的变化所引起,因此两者具有一定的正相关性[29]。本研究中,单核苷酸、二核苷酸重复次数相对较多,三核苷酸重复次数跨度相对较大,推测这3种核苷酸应具有更高的多态性。在基元长度方面,过滤掉长度<12 bp的片段后,草原1号杂花苜蓿花蕾转录组SSR长度主要集中在12~40 bp,长度在20 bp以上的SSR位点有9 166个,占比15.98%,这些多态性较高的SSR可能在杂花苜蓿中具有较高的应用价值。
本研究基于草原1号杂花苜蓿花蕾转录组的高通量测序数据,从总RNA水平上对特异性SSR位点进行了检索和序列特征分析,挖掘获得的SSR位点分布密度大、基元重复次数和类型丰富、多态性高,不仅为实用SSR标记的开发提供了数据,同时也有助于不同地区苜蓿的遗传多样性研究、苜蓿及其近缘种种质资源多态性分析、品种鉴定和分子标记辅助育种选择等相关工作的开展。
猜你喜欢
计算机工程与应用(2023年1期)2023-01-13
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
现代畜牧科技(2021年4期)2021-07-21
兵工学报(2021年4期)2021-06-19
中国饲料(2019年19期)2019-03-25
作文小学高年级(2019年1期)2019-02-18
科学导报(2018年30期)2018-05-14
中国三峡(2017年4期)2017-06-06
