
白细胞介素-37与牙周炎关系的研究进展
2021-08-25 12:20郑旭谢琛高畅郭竹玲
口腔疾病防治 2021年12期
郑旭,谢琛,高畅,郭竹玲
海南医学院第一附属医院口腔科,海南 海口(570102)
牙周炎为由牙菌斑导致的牙周组织炎症,可累及牙周膜、牙槽骨与牙骨质,其发生与发展和多种炎症因子有关。白细胞介素-1家族细胞因子大多为促炎因子,近年来研究发现,该家族的第七个成员IL-37具有强大的抗炎及抗自身免疫效果,成为目前的研究热点。现阶段较少关于IL-37与牙周炎相关因子的研究,本文将对两者进行综述。
1 IL-37 的结构及功能
IL-37共由218个氨基酸组成,其编码基因位于第二号染色体,可在胸腺、淋巴结、骨髓、肺部、胎盘、睾丸等部位表达。IL-37分为a~e共5个变异剪切体,IL-37b含有由第四号外显子负责编码的三叶草β状结构,对细胞因子和相应受体的结合具有重要作用。IL-37c、IL-37e无该结构,而IL-37a与IL-37d具有第四号外显子,故推测a、d亚型具有与IL-37b相似的功能。IL-37b具有caspase-1切割位点,可在该酶的作用下进出细胞核。实验表明,表达IL-37的转基因小鼠腹腔巨噬细胞对脂多糖诱导的促炎因子IL-1β、IL-6、TNF-α和IFN-γ的抑制率为40%~50%,当阻止caspase-1切割IL-37后,此抑制效果消失[1],说明IL-37可在该酶作用下发挥抗炎作用。IL-37在人类细胞和组织中低水平表达,但在炎症刺激和促炎细胞因子作用下表达上调[2]。IL-37具有强大的抗炎作用,分为细胞内和细胞外两种作用途径,在胞内可与Smads蛋白结合调控促炎基因转录,在细胞外IL-37可与IL-18结合蛋白(interleukin 18 binding protein,IL-18BP)或IL-1受体8(interleukin 1 receptor,IL-1R8)结合,抑制IL-18与IL-18Rα受体结合进而抑制炎症因子的产生,发挥抗炎作用[3]。此外,IL-37d亦可与Smads3结合抑制炎症因子的表达,而不是通过IL-18Rα/IL-1R8受体通路发挥抗炎作用[4](图1)。

Figure 1 The anti-inflammatory effects of IL-37 through intracellular and extracellular pathways图1 IL-37发挥抗炎作用的细胞内、细胞外途径
2 IL-37 与牙周炎相互关系研究
细菌微生物是牙周炎发生的始动因子,宿主的免疫反应在牙周组织破坏过程中发挥核心作用,树突状细胞、吞噬细胞等识别牙周组织中的病原微生物并将抗原呈递给T细胞、B细胞,引发自身免疫和适应性免疫应答,由树突状细胞提呈抗原、CD4+T细胞引发的牙槽骨吸收,以及破骨细胞的诱导形成和炎性因子释放在牙周炎病理过程中发挥重要作用。M1型巨噬细胞能够降低成骨基因Runt相关转录因子2(runt-related transcription factor 2,Runx2)表达水平以抑制成骨细胞分化,Th1细胞可通过促进破骨细胞形成加重牙槽骨的吸收程度[5]。记忆B淋巴细胞、骨细胞可通过高表达核因子-κB受体活化因子配体(receptor activator of NFκB ligand,RANKL)、增加骨硬化蛋白分泌、骨细胞凋亡及自噬的方式促进牙槽骨吸收,加重牙周炎进展[6-9]。IL-37可通过抑制RANKL途径、降低CD4+T细胞的活化程度抑制牙槽骨吸收,提示IL-37与牙周炎可能存在密切关联。研究发现,慢性牙周炎患者牙龈组织中IL-37水平明显高于健康组织,CD138+、CD38+浆细胞是慢性牙周炎牙龈组织中产生IL-37主要的免疫细胞类型[10]。IL-37可通过细胞内、细胞外途径抑制树突状细胞、CD4+T细胞增殖分化及炎症因子如IFN-γ、TNF-α的释放发挥抗炎作用。综上,提示IL-37与牙周炎发生发展存在联系。
2.1 IL-37与牙周炎相关细胞
2.1.1 CD4+T细胞 由CD4+T细胞引发的免疫反应在牙周炎发生发展中发挥重要作用,牙周炎患者牙龈产生局部超敏样反应,牙龈中特异性克隆CD4+T细胞增生活跃,导致大量IFN-α聚集于牙周炎患者牙龈中。在牙周炎症刺激下,CD4+T细胞可分化为多个亚型,Th1、Th17细胞表达增加,Th2细胞及Treg细胞表达水平显著下降,Th1细胞可通过分泌IFN-γ、IL-2、IL-12及RANKL导致牙周组织炎症扩散及牙槽骨的吸收,提示Th1细胞具有促进牙周炎进展的作用;Th2细胞分泌的细胞因子如IL-4具有高效的抗炎特性,Th2细胞的低水平表达可能与牙周炎的加重存在关联[11]。CD4+T细胞内有IL-37 mRNA产生,胞核周围可大量表达IL-37,T细胞内IL-37表达被沉默后,CD4+T细胞增殖活跃,促炎因子表达水平较对照组升高。有研究报道,Treg细胞在牙周炎组织中的含量高于正常牙周组织及牙龈炎组织,叉头框转录因子P3、转化生长因子-β(transforming growth factor-β,TGF-β)和IL-10表达水平较牙龈炎组织显著增高[12]。研究显示,Treg细胞内IL-37被沉默后,Treg细胞抑制免疫反应效果明显减弱[13]。在脂多糖存在条件下,IL-37对Treg细胞抑制T淋巴细胞增殖及促进TGF-β分泌的作用均加强。此外,Treg细胞在炎症因子作用下产生高水平的IL-37,IL-37可反作用于Treg细胞,提高其活性并促进抗炎因子TGF-β、IL-10的释放,IL-10能够抑制基质金属蛋白酶(matrix metalloproteinases,MMPs)产生和通过RANKL途径来缓解牙周组织的破坏程度。Treg细胞还可通过表达叉头框转录因子P3以降低牙周炎引发的自身免疫,延缓牙槽骨吸收的进程。此外,研究发现重组的IL-35和IL-37在体外对破骨细胞的形成具有剂量依赖性的抑制作用(在300 ng/mL的IL-35和IL-37中,对破骨细胞形成抑制率分别为78.9%和97.7%,P<0.05),推测牙龈组织中CD138+、CD38+浆细胞通过产生IL-35和IL-37,直接阻断破骨细胞的形成来抑制牙槽骨丢失,从而调节牙周炎的发展[10]。综上,IL-37通过抑制效应T细胞增殖和释放炎性细胞因子及作用于Treg细胞来抑制牙槽骨的破坏程度,发挥抑制牙周炎进展的作用。
2.1.2 树突状细胞 牙周炎牙龈组织中IL-37主要由牙龈上皮细胞和结缔组织中浸润的免疫细胞表达,有报道指出,牙龈组织的树突状细胞也是IL-37的来源之一[14]。未成熟状态树突状细胞具有抵御微生物侵入牙周组织的能力,在牙周抗原作用下促进树突状细胞成熟,表面共刺激分子CD80、CD86表达增加,进而激活CD4+T细胞、诱导Th1细胞增殖分化,促进牙周炎的发展[15]。慢性牙周炎患者血清中,树突状细胞共刺激分子CD80、CD86和CD40表达水平较牙周健康者显著升高,Th17/Treg比例上升,提示慢性牙周炎促进了树突状细胞成熟的过程[16]。树突状细胞在牙周炎状态下能够激活T细胞和天然免疫反应,促进CD4+T细胞分化成Th1、Th2、Th17和Treg细胞,导致基质金属蛋白酶和RANKL的表达,促进破骨细胞的形成和骨吸收,加重牙周炎的发生发展[16]。此外,未成熟的树突状细胞可以在牙周炎条件下参与破骨细胞的发生并导致骨丧失[17]。研究证明,IL-37预处理组树突状细胞的CD80、CD86表达水平明显下降,RANKL途径的表达亦受到抑制,提示IL-37可通过作用于RANKL途径而抑制树突状细胞的成熟[18]。当IL-1R8或TLR4缺乏时,IL-37对树突状细胞成熟的抑制作用明显减弱,提示IL-37通过IL-1R8-TLR4-RANKL途径来抑制树突状细胞成熟过程[19]。IL-37b可与细胞核内Smads蛋白结合,抑制树突状细胞的活化。加入适量浓度的IL-37在体外培养时,能够有效抑制树突状细胞表面产生共刺激因子。综上,IL-37可通过有效抑制树突状细胞的增殖而抑制牙周炎的进展(图2)。
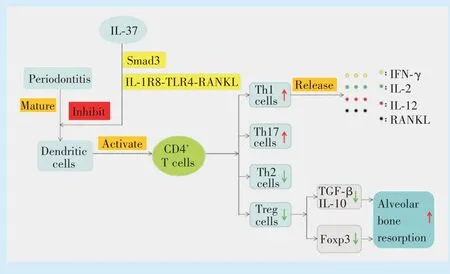
Figure 2 IL-37 inhibits proliferation and differentiation of dendritic cells and CD4+Tcells图2 IL-37抑制树突状细胞、CD4+T细胞增殖分化
2.2 IL-37与牙周炎相关促炎因子IFN-γ、TNF-α、IL-1β
IFN-γ是由辅助性T细胞产生的细胞因子,可以上调TNF-α和IL-1β的表达水平,刺激破骨细胞形成,加速牙槽骨的吸收进程。研究发现,IFN-γ在牙周组织炎症的扩散、牙周膜丧失及牙槽骨的吸收发挥重要作用[20]。在IL-37作用下,促炎因子如IFN-γ、TNF-α、IL-6表达水平明显降低,抑炎因子TGF-β表达显著升高[21]。
IL-37可以通过与IL-18BP结合,增强IL-18与IL-18BP结合的能力,抑制IL-18与IL-18Rα受体结合,进而抑制促炎因子IFN-γ产生,减缓牙周炎的发展[22]。
IL-1β可以促进MMPs、RANKL、前列腺素E2、IL-6、IL-8等的分泌,导致IL-1β具有强烈的骨吸收刺激作用,使其成为牙周炎发生发展的重要细胞因子。若阻断IL-37的正常表达,单核细胞在内毒素的作用下IL-1β表达增加2.1倍,提示IL-37可以通过抑制IL-1β的表达来抑制牙周炎症状态下牙槽骨的丢失[14]。研究表明,牙周炎患者唾液和血清TNF-α含量较牙周健康者高,TNF-α可上调牙龈上皮细胞、T细胞和成骨细胞中RANKL的表达,介导牙龈成纤维细胞和上皮细胞凋亡,抑制牙龈成纤维细胞产生细胞外基质,提示TNF-α可能通过破坏口腔黏膜屏障参与牙周炎的发生[23]。综上,IL-37可能通过抑制上述促炎因子的产生而发挥抑制牙周炎发展的作用。
2.3 IL-37与牙周炎的相关信号通路
2.3.1 RANKL途径 RANKL是破骨细胞的主要激活剂,活化的T细胞和B细胞是病变牙周组织RANKL的主要来源[24]。实验表明,选择性抑制RANKL/RANK轴可抑制牙槽骨的吸收[25]。在生理状态和炎症状态下,IL-37通过抑制RANKL途径的激活而抑制破骨细胞的分化,进而抑制牙槽骨吸收的进程[26]。骨细胞特异性RANKL缺失可完全阻断牙周炎,推测骨细胞表达的RANKL可能是牙周炎发生的重要环节[27],说明IL-37可通过作用于RANKL途径的激活而抑制牙周炎患者牙槽骨丧失。然而,有报道指出,IL-37可抑制LPS诱导的破骨细胞形成和骨吸收,并抑制LPS诱导的破骨细胞相关细胞因子的表达,但是IL-37对RANKL及TNFα诱导的破骨细胞形成无明显影响[28]。综上,IL-37是否可以作用于RANKL途径进而抑制牙周炎,还需更多相关实验进行验证。
2.3.2 Smads蛋白 Smads家族作为TGF-β家族的重要下游分子,与牙周炎症的发生发展存在密切关联。研究结果显示,Smads蛋白尤其是Smad3在人牙周膜干细胞骨向分化过程中起关键的信号转导作用[29]。Smad7蛋白可作用于免疫细胞的多种信号通路,促进大量炎症因子释放,还可与抑炎因子TGF-β竞争结合TGF-β受体,抑制细胞核中抑炎基因的表达。此外,Smad7蛋白还可增强RANKL途径,与牙周炎的发展存在密切关联。
免疫荧光结果显示,IL-37b可与Smad3结合形成复合物,影响基因的转录,抑制Toll样受体诱导的促炎细胞因子IL-1β、IL-16和IFN-γ的表达,发挥抑制炎症的作用[23],TLR信号在牙周炎症和骨吸收诱导中具有重要作用[30],Toll样受体可增强破骨细胞活性,促进牙槽骨的吸收。
细胞水平实验显示,前体IL-37b和成熟IL-37b均能与Smad3结合,干扰Smad2、Smad4与Smad3结合形成Smad2/3/4复合体,提示IL-37b可能通过与Smad2、Smad4竞争结合Smad3来抑制Smad通路[31]。IL-37和Smad3可形成功能复合物来抑制多种STATs的活性以及参与炎症信号通路的某些激酶的表达[32]。用特异性抑制剂SIS3阻断Smad3激活,IL-37抑制RAW细胞中促炎细胞因子的表达呈剂量依赖逆转现象,说明Smad3可能参与IL-37b的信号转导[3]。在小鼠RAW264.7细胞株和人THP-1细胞株中,使用Smad 3特异性抑制剂或靶向siRNA沉默内源性Smad 3后,IL-37的抗炎作用明显减弱。转染siRNA的IL-37转基因小鼠肺内Smad 3基因敲除也明显降低了IL-37的抗炎作用[32]。
研究发现,牙周炎患者牙周组织中Runx2表达水平较健康牙周组织显著降低,可能作用途径为Smad1/5/8磷酸化受到抑制,导致Runx2表达水平受限,进而抑制成骨细胞的分化[33]。在TGF-β存在下,IL-37显著增强了对Smad1/5/8磷酸化,而不是对Smad2/3的磷酸化[34],提示Smad1/5/8磷酸化可能为IL-37抑制牙周炎进展的途径之一。综上,Smads蛋白可能在IL-37抑制牙周炎过程中起重要作用(图3)。
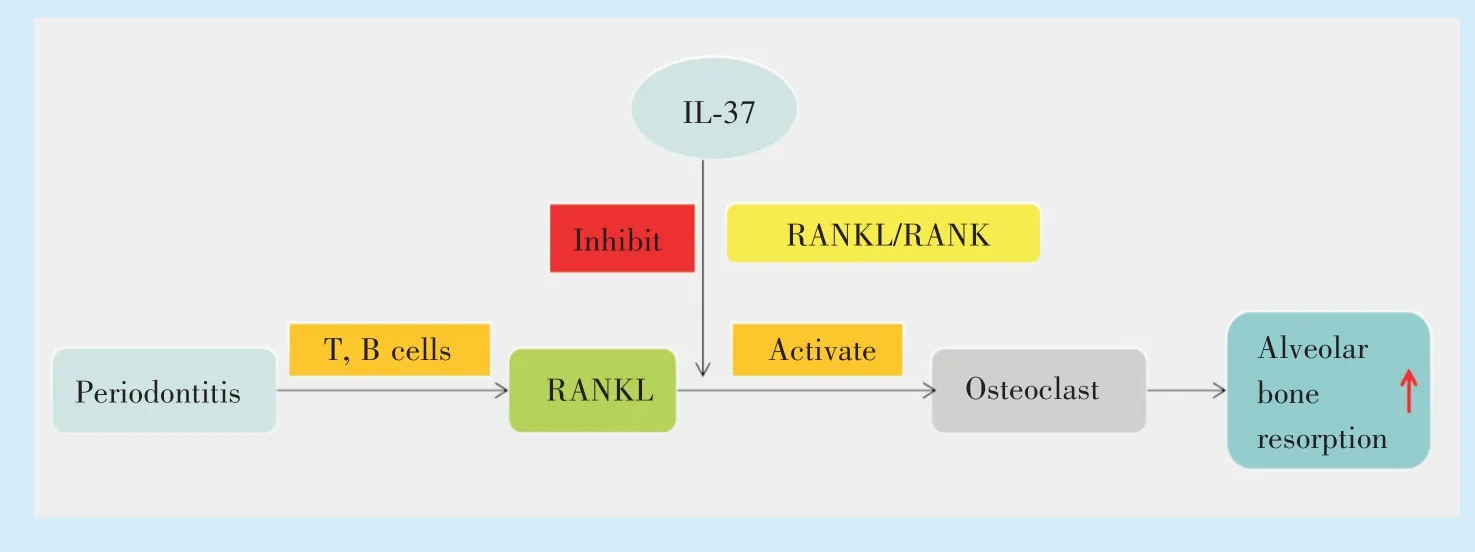
Figure 3 IL-37 delays alveolar bone resorption through inhibiting the RANKL pathway图3 IL-37通过抑制RANKL途径延缓牙槽骨吸收
3 小结
作为IL-1家族的新成员,IL-37可通过细胞内和细胞外两种途径发挥抑制炎症发展的作用。许多临床试验表明,牙周炎患者龈沟液中IL-37的浓度具有明显变化,作为牙周组织的炎症疾病,IL-37可能通过作用于CD4+T细胞、Treg细胞,抑制树突状细胞增殖,与Smads蛋白结合和抑制RANKL途径发挥抑制牙周炎进展的作用。牙周炎的发生机制尚未阐明,IL-37或许可成为牙周炎发生发展机制的关键抑制因子,为牙周炎治疗的研究提供新的思路,现阶段仍需更多相关研究加以验证。
【Author contributions】 Zheng X wrote the article.Xie C,Guo ZL,Gao C revised the paper.All authors read and approved the final manuscript as submitted.
猜你喜欢
中国临床解剖学杂志(2022年3期)2022-06-06
锦州医科大学报(2022年2期)2022-05-07
今日健康(2021年10期)2021-12-01
河南大学学报(医学版)(2021年1期)2021-11-26
海外星云(2021年6期)2021-10-14
昆明医科大学学报(2021年5期)2021-07-22
中华养生保健(2020年7期)2020-11-16
中华养生保健(2020年3期)2020-11-16
中西医结合肝病杂志(2020年2期)2020-10-27
数理医药学杂志(2020年10期)2020-10-17
