
cGAS-STING信号通路和自身免疫性疾病①
2021-08-21 05:19:58俞宛君江苏大学医学院免疫学与免疫检验学教研室镇江212013
中国免疫学杂志 2021年14期
俞宛君 夏 圣(江苏大学医学院免疫学与免疫检验学教研室,镇江212013)
自身免疫性疾病(autoimmune disease,AID)是一组由于自身免疫耐受被打破而引起的组织、器官慢性炎症性疾病,以自身反应性T细胞、B细胞过度活化、自身抗体大量产生为基本特征[1]。AID可分为器官特异性AID和系统性AID,其中,器官特异性AID常见的有桥本氏甲状腺炎、胰岛素依赖型糖尿病、慢性溃疡性结肠炎等;而系统性AID最常见的是系统性红斑狼疮(systemic lupus erythematosus,SLE)。SLE的免疫异常几乎覆盖整个免疫系统,被认为是AID的原型,因此人们对SLE的发病机制研究也更为深入。研究发现,天然免疫细胞产生的Ⅰ型干扰素(typeⅠinterferons,IFN-Ⅰ)在全身自身免疫中起核心作用,可激活B细胞和T细胞。反之,B细胞产生的自身抗体也可刺激树突状细胞产生IFN-Ⅰ,从而形成一个包括天然免疫和适应性免疫的正向反馈回路,即当机体自身免疫平衡被打破,胞质DNA持续或过度存在引起IFN-Ⅰ持续产生是导致慢性炎症和AID的主要原因之一[2]。因此,新的治疗方式或许可以同时将针对介导自身免疫的多个重要通路作为靶点[3-4]。
目前,临床上依然缺乏关于AID的理想治疗方案。当前方案通常仅针对疾病的病理性组织损伤等进行治疗,同时对患者的免疫系统进行抑制。此法或许能够阻断疾病的继续恶化,但长期治疗可并发感染、诱发肿瘤和骨髓抑制等不良后果或副作用。近年来,研究者们也尝试通过作用不同靶点,包括靶向不同细胞因子、不同淋巴细胞表面标志及合成肽免疫原等生物制剂,调节免疫应答的各个环节,阻碍疾病进程[5]。由于对AID发病机制的研究尚未清楚,各类生物制剂的疗效及安全性还需大规模多中心双盲随机对照试验进行验证。随着全球AID发病率逐年升高,尽快明确AID的发病机制,并探索合理有效的治疗方案已成为临床的迫切需求。
1 cGAS-STING信号通路
1.1 cGAS-STING信号通路概述 天然免疫是机体与生俱来的防御能力。天然免疫系统通过模式识别受体(pattern-recognition receptors,PRRs)识别病原相关分子模式(pathogen associated molecular patterns,PAMPs)和损伤相关分子模式(damage-associated molecular patterns,DAMPs),迅速对外来入侵的病原体以及自身坏死、凋亡或受损组织产生免疫应答,筑起保护机体的第一道防线[6-7]。能够识别细胞内核苷酸的PRRs有维甲酸诱导基因Ⅰ样受体(RIG-Ⅰlike receptors,RLRs)、核苷酸结合结构域NOD样受体(NOD-like receptors,NLRs)、黑色素瘤缺乏因子2(absentinmelanoma 2,AIM2)、干扰素诱导蛋白16(interferon-inducible protein 16,IFI16)以及近年新发现的cGAS-STING信号通路相关分子等。
cGAS-STING信号通路的信号传递,主要分为dsDNA的感知、胞内信号转导和免疫应答激活3个阶段。dsDNA感知阶段主要包括以环鸟苷酸-腺苷酸合成酶(cyclic GMP-AMP synthase,cGAS)为首的一系列分子;胞内信号转导分子主要有干扰素基因刺激蛋白(stimulator of interferon gene,STING)和TANK结合激酶1(TANK-binding kinase 1,TBK1);而免疫应答激活由多种免疫应答相关转录因子介导下游靶基因的表达,如干扰素调节因子3(interferon regulatory factor 3,IRF3)和核因子NF-κB(nuclear factor kappa-light-chain-enhancer of activated B cells)。
cGAS存在于胞浆中,是一种核酸转移酶,含有一个与寡腺苷酸合成酶(oligoadenylate synthase,OAS)催化结构域同源的结构域,依靠其氨基端DNA结合位点和羧基端锌指结构,识别dsDNA磷酸-核糖骨架结构,即几乎能够识别所有类型的dsDNA[8]。相 较 于 其 他 识 别 细 胞 质 内DNA的PRRs,cGAS具有更强的激活效应,且其在多种组织和细胞中都有表达,被认为是胞浆DNA诱导干扰素产生的主要信号通路分子[9]。cGAS识别并结合dsDNA后,催化ATP和GTP生成环磷酸鸟苷-磷酸腺苷(cyclic guanosine monophosphate-adenosine monophosphate,cGAMP)。2013年GAO等[10]和ABLASSER等[11]分别利用质谱、酶切、核磁共振分析和化学合成等方法揭示cGAS在体内和体外催化合成的二核苷酸不同于细菌产生的经典3'-5'-环二核苷酸,是含有非经典的2'-5'磷酸二酯键结构c[G(2',5')pA(3',5')p]的环二核苷酸(cyclic dinucleotides,CDNs)。有研究利用反相高效液相色谱技术发现c[G(2',5')pA(3',5')p]分两步合成。首先,形成2'-5'磷酸二酯键连接,而后再形成3'-5'磷酸二酯键连接,称为2'5'-cGAMP。与3'5'-cGAMP相比,2'5'-cGAMP对于人体细胞内STING蛋白亲和力高,可诱导高水平的干扰素诱导蛋白10(interferon inducible protein-10,IP-10)[10-11]。
STING又称跨膜蛋白173(TMEM173)、MITA等,是一种跨膜接头蛋白,主要在巨噬细胞、树突状细胞、T细胞等的内质网中普遍表达,是异常胞浆DNA非特异性免疫的重要组成部分。研究发现STING可直接结合dsDNA,也可被其上游含有两个3'-5'磷酸二酯键的原核CDNs及2'5'-cGAMP诱导活化,导致STING构象发生变化[12]。活化的STING可募集TBK1形成STING复合物。该复合物可从内质网区域通过高尔基体快速转运,定位于近核小体,而后结合核转录因子IRF3,并诱导后者磷酸化和二聚化后进入细胞核。STING亦可激活IKK(inhibitor of nuclear factor kappa-B kinase),导致NF-κB被释放并转位至细胞核内。在细胞核内IRF3及NFκB两种转录因子激活相关基因的表达,诱导IFN-Ⅰ及相关细胞因子的产生。STING随后发生磷酸化、泛素化等修饰,活性受到抑制,防止天然免疫反应的过度激活(图1)[13]。
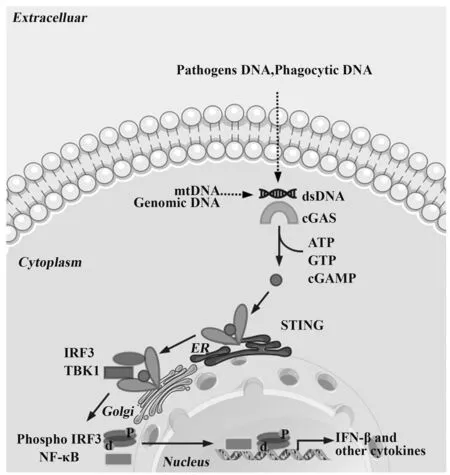
图1 cGAS-STING信号通路示意图Fig.1 Schematic diagram of intracellular cGAS-STING signal pathway
1.2 cGAS-STING信号通路调控 当病原微生物感染人体时,病原体外源DNA能够被机体胞质核酸受体cGAS识别,利用细胞内的ATP和GTP生成第二信使cGAMP。cGAMP能够和内质网定位蛋白STING相互作用,导致STING构象变化,从而招募并磷酸化激活TBK1和IKK,继而分别激活IRF3和NFκB。而IRF3则进入细胞核,促进IFN-Ⅰ表达,启动天然免疫反应抵御病原菌入侵[14]。cGAS除了识别外源DNA外,细胞质内自身DNA的异常累积同样可被cGAS辨别,从而持续性激活cGAS-STING信号通路,导致机体AID的发生[15-18]。
为维持免疫自稳,cGAS-STING信号通路在机体内必然受到精密的调控。当前的研究主要集中在cGAS和STING蛋白翻译后修饰(磷酸化、泛素化、苏木化及谷氨酰化等)、cGAS和STING蛋白自身突变及信号通路交互作用等方面。此外,离子环境对cGAS-STING信号通路也有调节作用,如Mn2+可增加cGAS及STING活性,帮助机体抵御DNA病毒感染[19]。AKT激酶分别磷酸化小鼠和人cGAS结构域的S291和S305残基,强烈抑制其酶活性,在cGAS介导的抗病毒免疫反应中起负作用[20]。E3泛素连接酶RNF185(ring finger protein 185,RNF185)特异性催化cGAS的K27多泛素化,提高其酶活性,同时SLE患者RNF185 mRNA表达升高,但对于RNF185在SLE中发挥的具体功能还需进一步探索[21]。泛素羧基末端水解酶27(ubiquitin carboxyl-terminal hydrolase27,USP27x)可与cGAS相互作用,并从cGAS上解离K48连接的多泛素化链,从而使cGAS稳定[22]。泛素连接酶TRIM56(tripartite-motif-containing protein 56,TRIM56)诱导cGAS的Lys335位点单泛素化,使其二聚化、DNA结合活性和cGAMP产量显著增加[23]。类微管蛋白酪氨酸连接酶6(tubulin tyrosine ligase-like enzymes 6,TTLL6)多聚谷氨酰化cGAS抑制其与DNA的结合能力,而类微管蛋白酪氨酸连接酶4(TTLL4)介导的cGAS单谷氨酰化则抑制其合酶活性。相反,羧肽酶6(cytosolic carboxypeptidase 6,CCP6)去除了cGAS的多谷氨酰化,而羧肽酶5(CCP5)水解cGAS的单谷氨酰化,共同导致了cGAS的激活[24]。在未感染及病毒感染早期阶段的细胞中,泛素连接酶TRIM38(tripartite-motif-containing protein 38,TRIM38)以cGAS为靶标进行苏木化,可阻止cGAS多泛素化和降解[25]。STING作为胞内核酸识别信号通路关键接头蛋白,其受到调控的研究更为深入。STING的Ser366磷酸化修饰功能存在争议。有研究表明,TBK1磷酸化STING后可促进IRF3的磷酸化二聚化并入核[26],而激酶ULK1(unc-51 like autophagy activating kinase 1,ULK1)磷酸化STING相同位点后却抑制STING活性[13],因此还需进一步实验确证。线粒体E3泛素连接酶1(mitochondrial E3 ubiquitin protein ligase 1,MUL1)通过催化STING蛋白K224泛素化促进TBK1向IRF3的信号传递[27]。内质网E3泛素连接酶复合体自分泌运动因子受体-胰岛素诱导基因1(autocrine motility factor receptor-insulin induced gene 1,AMFR-INSIG1)催化STING蛋白K27多泛素化来招募TBK1,并促进其转位到核周[28]。而泛素连接酶29(tripartite-motifcontaining protein 29,TRIM29)靶向STING蛋白K48泛素化,从而降解STING[29]。需要注意的是,对于cGAS和STING蛋白翻译后修饰的研究主要集中于抗病毒免疫领域,其在自身炎症和AID中对于cGAS-STING信号通路的调控需要进一步实验验证。有趣的是,高尔基体中STING的棕榈酰化对于其激活至关重要。抑制STING棕榈酰化可抑制SAVI相关的STING突变体引起的IFN-Ⅰ反应[30]。这可能为胞浆DNA引发的自身炎症和AID提供新的治疗方法。
2 自身DNA与cGAS-STING信号通路
人体正常细胞的DNA位于细胞核或线粒体细胞器内,而细胞质内则不含DNA。因此,细胞质中若有DNA存在,一方面可能源自入侵的细菌性病原体或DNA病毒;另一方面可能源自细胞核或线粒体遭受的损伤。cGAS作为感知DNA的监测器,可提示先天性免疫系统存在的威胁。而关于机体如何避免各类PRRs不适当地识别内源性DNA,其可能机制有:①宿主受体可能优先检测病原体基因组结构,如细菌CpG基序;②宿主受体可能被分配到一个有限的、没有自身DNA的亚细胞区域;③许多内源性核酸的水平低于受体激活阈值[31];④对未经氧化修饰自身DNA的免疫监测低于由抗菌活性氧或物理损伤引起的氧化DNA,从而避免自身免疫反应发生[32]。同样,cGAS作为一种不依赖于dsDNA序列的通用胞质内DNA感受器,在固有免疫中也发挥“双刃剑”的效应,既要减少对自身DNA的识别,又要识别外源病毒的DNA。有研究分析小鼠cGASDNA复合物晶体结构,发现长链DNA可结合更多的cGAS二聚体而增强酶活性[33]。随后,陈志坚课题组报道了cGAS-DNA的相分离现象,发现较长的DNA片段比短DNA片段更能促进液滴形成,且相变对DNA浓度敏感,证实cGAS的激活具有DNA长度和浓度的依赖性[34]。在此基础上,DINSHAW团队通过分析人类cGAS-DNA复合物晶体结构揭示人源cGAS催化结构域有一个额外的DNA结合界面,可增强酶活性和液相缩合[35]。这些发现部分解释了cGAS执行胞内免疫监视功能时规避碎片DNA及低浓度DNA干扰的机制。最近,哈佛大学JONATHAN团队的研究指出,cGAS并非既往理解的胞质蛋白,而是通过其N端结构域与质膜脂质结合的膜定位蛋白。在静息状态下,cGAS的N端的胞膜定位可防止cGAS移位于胞质,避免其识别胞质内微量DNA,从而减少活化;而当cGAS异常定位时,并不增加其对外源病毒感染的免疫效应[36]。而自身DNA也可通过如下所述的多种机制激活cGAS-STING信号通路。
2.1 自身DNA清除缺陷3'-5'核酸修复外切酶1(three-prime repair exonuclease 1,TREX1)是一种细胞内非常重要的内源性DNA核酸外切酶,可通过水解受损的DNA来清除细胞内需要处理的DNA片段,从而避免过度免疫激活和AID的发生。研究人员发现,Trex1-/-小鼠可于出生后第8周左右发生致死性炎症性疾病。若同时敲除Irf3,可有效避免高水平的IFN-Ⅰ及自身抗体的产生,防止Trex1-/-小鼠早期死亡,此结果说明Trex1-/-动物的自身免疫症状与IRF3依赖的IFN-I产生有关[37]。同时,Trex1-/-小鼠表现出与干扰素刺激基因(IFN-stimualted genes,ISGs)表达升高相关的自身免疫炎症表型。而Trex1-/-Cgas-/-小鼠所有可检测到的病理表现和分子表型都得到缓解,包括ISGs诱导、自身抗体产生、异常T细胞激活和致死性。即使只缺失1个Cgas等位基因,也能在很大程度上挽救Trex1-/-小鼠的表型。这些结果提示,抑制cGAS可能有助于防治某些自身DNA引起的AID[38]。另有研究发现,TREX1对于氧化损伤的DNA分子降解效率较低,在易发生狼疮的小鼠皮肤中注射氧化DNA可引起与AID患者类似的病变[32]。TREX1功能缺失突变会引起IFN-Ⅰ依赖性AID,如AGS综合症(Aicardi-Goutieres syndrome,AGS)和家族性冻疮样狼疮(familial chilblain lupus,FCL)等,这些自身免疫紊乱的表现都与细胞无法正常清除产生的自身DNA片段有关[39-40]。
脱氧核糖核酸酶Ⅱ(deoxyribonucleaseⅡ,DNaseⅡ)是位于溶酶体、细胞核以及细胞分泌物中的一种酸性核酸内切酶。DNaseⅡ家族在人体内参与多种功能,如降解被吞噬死亡细胞的DNA,参与红细胞的生长分化等。DnaseⅡ-/-小鼠吞噬细胞功能遭到破坏,可引起严重的免疫缺陷并影响生长发育[41]。有报道称,DnaseⅡ-/-小鼠在子宫内胚胎期即死亡,该小鼠胚胎肝脏中含有许多未消化DNA的巨噬细胞,表明DnaseⅡ-/-小鼠不能降解从红系前体细胞衍生的DNA,导致IFN-I的产生,诱导特定ISGs的表达,导致DnaseⅡ-/-小鼠胚胎期致死[15,38,42]。而成年期的DnaseⅡ-/-鼠可死于类似于类风湿性关节炎的慢性多关节炎[43]。对于DnaseⅡ-/-Cgas-/-小鼠而言,该小鼠可正常发育,无明显的关节炎症状,抗ds-DNA抗体也呈低水平,并且DnaseII-/-Cgas-/-小鼠中未检测到cGAMP表达,而在DnaseⅡ-/-Sting-/-小鼠中cGAMP高表达。因此,可认为针对胞浆中累积自身DNA的应答,cGAS对DnaseⅡ-/-细胞中cGAMP的产生是必不可少的[38]。有研究通过对ISGs的系统筛选,确定了患有严重新生儿贫血、膜增生性肾小球肾炎、肝纤维化的两兄妹和一个单独男性病例,均伴随抗DNA抗体的升高。在这两个家族中,发现DNASE的双等位基因突变,与DNaseⅡ内切酶活性的丧失有关[44]。
核糖核酸酶H2(RNase H2)是一种核酸修复酶,可从DNA中清除不必要的核糖核苷酸。RNase H2酶功能的改变是儿童AGS综合症的常见原因,与SLE也有关[45]。有研究利用Rnaseh2bA174T/A174T小鼠作为亚临床疾病模型研究发现,该小鼠体内ISGs转录上调,与AGS患者特征相似,此炎症反应依赖于核酸传感器cGAS及其适配器STING,并与细胞RNase H2酶活性降低和DNA损伤增加有关[46-47]。Cgas-/-或Sting-/-可挽救Rnaseh2bA174T/A174T小鼠的炎症和自身免疫表型[46]。
2.2 自身DNA泄漏
2.2.1 线粒体DNA应激 线粒体DNA(mitochondrial DNA,mtDNA)在胞内通常以数千拷贝的形式存在,并被包装成几百个称为“类核”的高阶结构。线粒体转录因子A(mitochondrial transcription factor A,TFAM)与丰富的mtDNA结合调节“类核”的结构、丰度和分离。有研究显示,由TFAM缺乏引起mtDNA应激,异常的mtDNA可逃逸到胞浆中,与DNA传感器cGAS结合,并促进STING-IRF3依赖信号,以提高ISGs表达,增强IFN-Ⅰ反应和广泛的病毒抗性[48]。也有研究表明,细胞死亡的机制决定了死亡细胞是触发炎症反应还是保持免疫沉默,而线粒体在诱导细胞死亡以及免疫沉默信号通路中起核心作用。在没有含半胱氨酸的天冬氨酸蛋白水解酶(cysteinyl aspartate specific proteinase,caspase)的情况下,线粒体外膜通透性增高可通过mtDNA依赖激活的cGAS-STING途径介导IFN-Ⅰ表达[49]。YU等[50]发现在非酒精性脂肪性肝炎小鼠模型中,小鼠肝细胞内mtDNA可诱导肝脏Kupffer细胞表达TNF-α和IL-6,将小鼠Sting基因敲除或使用NF-κB抑制剂预处理后,mtDNA诱导炎症因子表达的作用减弱。值得注意的是,SLE中也存在功能失调的线粒体。有研究发现,SLE患者血浆白细胞mtDNA拷贝数的减少与SLE疾病活动性指数的升高和血浆DNA氧化损伤标志物8-羟基脱氧鸟苷水平的升高相关[51]。
2.2.2 细胞核DNA染色质传统上被认为是一个调节基因表达和沉默的核实体。然而,最近研究发现了由原代细胞的完整细胞核在衰老过程中剥离出来的细胞质染色质片段的存在。这是一种与促炎反应相关的终末细胞周期阻滞的形式,但染色质在细胞质中的功能意义尚不清楚。有研究发现,细胞质染色质可激活天然免疫胞质DNA传感cGASSTING途径,在急性炎症中抑制原癌基因激活,但在慢性炎症中与组织损害和癌症有关。细胞质染色质-cGAS-STING通路促进原代人细胞和小鼠衰老相关分泌表型。Sting-/-小鼠对癌基因Ras的免疫监测受损,对电离辐射导致的组织炎症反应减弱[52]。另有研究报道,在共济失调毛细血管扩张症患者中,DNA损伤修复受损促进细胞核DNA释放并在细胞质中积累,导致IFN-Ⅰ产生,同时导致患者中性粒细胞中自发形成胞外诱捕网[53]。2017年德克萨斯大学西南医学中心YANG等[54]的研究显示,cGAS定位于非分裂细胞的细胞质,但当增殖细胞处于有丝分裂时期,cGAS可进入细胞核并与染色质DNA结合。cGAS核定位对于其执行其抗病毒功能可能是必要的,因为大多数DNA病毒感染宿主细胞后会进入细胞核内,释放病毒基因组DNA并在核内进行复制。随后,有研究发现cGAS在细胞发生DNA损伤时可转位入核,并在DNA损伤位点抑制DNA同源重组修复,进而降低基因组稳定性,促进肿瘤发生,这独立于cGAS核内DNA识别的功能[55]。因此,cGAS核转位机制及其能否区分细胞核内的自体和非我DNA仍有待进一步研究。癌细胞普遍具有染色体不稳定性。BAKHOUM等[56]的研究发现,人体发生癌症转移的部分原因可能与体内肿瘤细胞DNA慢性泄露有关。泄露DNA激活cGAS-STING通路,从而引发细胞内的慢性炎症,帮助肿瘤细胞向远处器官扩散。
以上研究表明,cGAS-STING通路在自身炎症和AID发展进程发挥重要作用。若适当抑制该通路活性,会减缓AID疾病的进程。
3 cGAS-STING信号通路与AID的诊断和治疗
通常情况下,炎症反应有助于消除感染,但发生在某些疾病中的持久炎症却能够损伤机体。cGAS-STING途径的不当激活或过度激活可产生大量IFN-Ⅰ,释放至细胞外的IFN-Ⅰ能够以自分泌或旁分泌的方式,结合自身或者周围细胞上的IFN-Ⅰ受体(IFNα receptor,IFNAR),进一步激活其下游JAK-STAT通路,诱导数百个ISGs的表达,导致自身炎症和AID[57-58]。
一些研究表明,AID与cGAS-STING通路中相关蛋白编码基因突变相关。因此,可通过基因检测辅助诊断AID。2014年的《新英格兰医学杂志》刊登了6例STING相关婴儿期起病的血管病(sting-associated vasculopathy with onset in infancy,SAVI)。这些病患均由于STING编码基因TMEMl73获得性突变所致,p.V155M为该基因的热点突变。该病可散发,亦可在家系中呈常染色体显性遗传模式,其病例特点包括新生儿期全身炎症反应,严重的组织坏死丢失和间质性肺病[59]。2018年中国医学科学院及北京协和医学院共同报道中国大陆地区首例SAVI病例的临床表现及基因突变位点[60]。连同干扰素通路激活的其他证据表明,TMEMl73突变可增强STING蛋白活性,STING高水平地存在于血管内皮细胞和肺部,可导致皮肤血管炎症发生,进一步发展为大范围肺组织损伤。STING的过度激活可产生大量IFN-Ⅰ,当其与IFNAR结合后,经过JAK-STAT信号通路进一步级联活化,产生炎症因子风暴效应,从而对STING高表达的组织造成严重持久损伤[61-63]。FCL是一种由于核酸酶TREX1或SAMHD1功能缺失突变引起单基因皮肤红斑狼疮。在一个没有TREX1或SAMHD1突变的多代FCL家系中,研究者们发现了STING的杂合突变。结构和功能分析表明,突变的STING在没有配体cGAMP的情况下促进了其同源二聚作用,从而导致结构性的IFN-Ⅰ激活[64-65]。
近年来,人们发现许多潜在靶向药物和分子可用于治疗由cGAS-STING通路异常引发的疾病。HAAG等[66]报道了靶向STING蛋白的高效特异性小分子抑制剂C-176、C-178以及H-151。这些小分子可与人和小鼠细胞STING蛋白中的Cys91发生共价结合,阻断STING活化所诱导的棕榈酰化,阻碍STING多聚体复合物形成,抑制下游免疫应答激活,减弱小鼠自身炎症疾病的病理特征。该类化合物活性强且分子量低,药理实验结果显示能够有效阻断机体天然免疫反应的信号通路,是极具潜力的靶向治疗cGAS-STING通路相关AID的候选药物。2020年1月,Dr.NAN团队研究发现了维持STING蛋白稳态的一个关键蛋白TOLLIP。敲减Tollip基因可导致非造血细胞和组织中STING蛋白表达减少,并使免疫细胞中的STING蛋白不稳定,导致STING信号转导能力严重减弱。Tollip-/-可改善STING介导的Trex1-/-小鼠的自身免疫炎症表型。因此,TOLLIP可以作为一个潜在的靶标用于治疗cGAS-STING通路相关的AID[67]。国内也有研究发现,小分子环肽astin C可选择性地抑制胞质DNA所诱发的炎症反应。HSV-1感染Trex1-/-小鼠时,astin C可阻断IRF3被招募到STING信号小体,从而显著降低其自身免疫炎症反应[68]。在AGS小鼠模型中,作为cGAS与DNA相互作用抑制剂的化合物X6疗效优于羟氯喹,可显著降低小鼠脾脏细胞ISGs的表达。值得关注的是,X6在降低SLE患者外周血单个核细胞(peripheral blood mononuclear cells,PBMCs)ISGs表达方面也明显优于羟氯喹。此结果表明,X6可作为AGS和SLE的一种新的治疗方法来抑制cGAS-STING通路的激活[69]。阿司匹林可使cGAS发生乙酰化从而抑制cGAS活性。阿司匹林可显著抑制AGS综合征模型Trex1-/-鼠心脏中ISGs的表达,并有效延长小鼠的生存。针对国内利用一例AGS综合征患者及其健康的亲兄长的PBMCs研究显示,阿司匹林处理可明显抑制该患者PBMCs中过度免疫反应表型[70]。值得注意的是,在疾病的病理生理过程中,细胞通常会接受多种刺激,或者同时经历许多环境变化。因此,在这种更复杂的环境中,cGAS-STING激活的细胞反应可能不同。IFN-Ⅰ在SLE发病机制中占有重要地位,KATO等[71]研究发现SLE患者血清中的凋亡衍生膜泡可通过激活cGAS-STING途径诱导IFN-Ⅰ产生。此结果证实了DING等[18]的假设,即多个器官中cGAS-STING通路对不适当的胞浆自身DNA感知来促进SLE的整体疾病进展。然而,也有研究报道STING有效地抑制了狼疮易感模型小鼠的炎症,进一步研究发现Sting-/-可能通过增加Toll样受体的表达水平或减少Toll样受体信号通路的负调控,从而导致巨噬细胞的放大激活效应[72]。因此,在探索cGAS或STING作为靶标进行AID治疗的过程中需要注意治疗所导致的其他效应。
4 展望
自2013年首先鉴定cGAS是细胞质DNA免疫识别通路的DNA受体以来,cGAS-STING信号通路成为国内外研究的热点领域。近年来,许多研究通过对通路关键分子的深入探讨,逐渐解开了当机体遇到细菌或病毒时,宿主细胞如何通过感知病原体核酸来诱导IFN-Ⅰ抵御侵袭。而本综述简要阐述了自身DNA触发cGAS-STING信号通路机制及靶向此通路自身炎症和AID的潜力药物和分子。事实上,cGAS及STING抑制剂被认为具有广泛的治疗潜力,可作为备选来补充现有的治疗策略,甚至作为单独用药的治疗方案。cGAS及STING抑制剂不仅对AGS、FCL、SAVI及SLE等自身炎症和AID有治疗的探索价值,而且对更多因异常cGAS-STING通路激活诱发的常见疾病,如非酒精性脂肪性肝炎、慢性阻塞性肺疾病和帕金森病等也有潜在治疗价值。前述靶向药物多停留在动物实验阶段并在动物模型中显示出良好的治疗效果,但进一步的试验可能因非靶标效应而受到阻碍,如增加机会感染的风险等。值得注意的是,从理论而言,与现有阻断IFN-Ⅰ下游信号通路(如JAK抑制剂)的治疗药物相比,cGAS及STING抑制剂可能更具优势,因为其他识别细胞质内核苷酸的信号通路相对完整。另外,对cGAS-STING通路的负向调控还可减少其他炎症因子如IL-6、TNF-α表达,减轻细胞因子风暴及组织损伤程度。cGAS-STING通路更详细的分子机制正在逐渐被解析,但许多问题仍然存在。如大多数研究都使用了小鼠模型和细胞系,但两者之间有很大的区别,且在人类的临床试验中更是受到巨大挑战。因此,继续深入了解STING激活的分子信号机制,将会推进针对感染、癌症和AID新治疗策略的开发。
猜你喜欢
数学物理学报(2021年4期)2021-08-30 08:27:48
新世纪智能(数学备考)(2020年10期)2021-01-04 00:37:50
中成药(2017年12期)2018-01-19 02:06:52
中国交通信息化(2017年8期)2017-06-06 07:16:47
中国病理生理杂志(2015年8期)2015-12-21 12:38:06
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15 12:47:42
医学研究杂志(2015年3期)2015-06-10 06:41:52
中国医学科学院学报(2015年5期)2015-03-01 04:03:46
创业家(2015年1期)2015-02-27 07:52:02
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
