
全氟己酸迁移反应机理的理论研究
2021-08-10 08:27宣敏敏刘子忠郭利超赵晓霞许天孜
内蒙古师范大学学报(自然科学汉文版) 2021年4期
宣敏敏, 刘子忠,3, 郭利超, 赵晓霞, 许天孜
(1.内蒙古师范大学 化学与环境科学学院,内蒙古 呼和浩特 010022;2.内蒙古自治区绿色催化重点实验室,内蒙古 呼和浩特 010022;3.内蒙古师范大学 应用数学中心,内蒙古 呼和浩特 010022)
自20世纪40年代初,全氟羧酸(PFCAs)化合物就因其良好的耐热性、高表面活性及化学稳定性等特点[1-2],作为灭火剂、乳化剂、吸附剂等广泛应用于工业和日用品中[3-4]。PFCAs作为一种持久性有机污染物[5],因其在环境中的大量存在和对人体潜在的健康影响[6-7],引起广泛关注[8-9]。
人们开始对自然界中PFCAs的含量以及种类进行检测分析[10],在中国氟化工厂附近流域的地表水中,检测到直链PFCAs的含量约为58%,支链PFCAs约为42%[11]。日本海和Tomakomai Bay中支链全氟辛酸(PFOA)比例分别为15%和16%[12]。美国Ontario Lake水样中,支链PFCAs比例在5.1%~6.0%之间。加拿大Nunavut、Char Lake和Amituk Lake所采水样中,支链PFCAs含量也很低[13]。Fang等[14]通过实验分析得到空气颗粒物中,直链的PFOA也占主要组成部分,支链异构体仅占10%。通过对自然界中的PFCAs的含量以及种类检测分析可知,自然界的河流、土壤甚至大气中既有直链也有支链的PFCAs,但任何环境中直链的PFCAs含量均大于支链[15]。实验制备过程中直链与支链的比例与自然界中检测结果一致,在使用电化学氟化法生产PFCAs时,直链的PFCAs约占70%,支链异构体约占30%[16]。2019年,Liu等[17]通过量子化学计算研究发现,在C原子数相同下,支链PFCAs比直链PFCAs能量低,根据能量最低原理,支链结构更能稳定的存在,理论预测自然界水体中支链PFCAs的含量应高于直链结构。对比理论和实测结果,发现二者明显存在矛盾。探究其不一致的原因也引起高度关注。
当前市场使用的大部分PFCAs均来自电解工业生产所制的工业产品,工业生产的PFCAs均是直链和支链异构体的混合物[18],且直链结构含量明显高于支链结构含量,使用后直接排放到水体,导致自然界水体中直链结构含量高于支链结构含量。为了探究直链结构能否向支链转化这一问题,以直链全氟己酸(PFHxA)为代表,运用Gaussian 09计算软件,对直链PFHxA中单个和两个全氟甲基迁移生成等碳原子的支链全氟戊酸和全氟丁酸的6种可能迁移途径进行过渡态优化,活化能和反应速率计算,进而找出直链PFHxA 迁移生成支链PFCAs的最佳迁移路径,明确直链全氟羧酸迁移生成支链结构的反应机理,揭开自然界水体中高能的直链结构含量高于支链结构内在原因,为进一步探究全氟羧酸的降解提供理论参考。
1 理论模型与计算方法
所有计算均在Gaussian 09程序中完成。在B3LYP/6-31G(d,p)计算水平下,对气相和水溶剂中的PFHxA通过全氟甲基迁移反应生成等碳原子的支链全氟戊酸和全氟丁酸的6种可能迁移途径中所涉及的各种反应物、产物、中间体和过渡态的构型进行全参数优化,同时进行振动频率分析,对每一个过渡态进行内禀反应坐标(IRC)分析,验证各过渡态与相应反应物和产物(中间体)连接的正确性。在B3LYP/6-31G(d,p)计算水平下,计算了反应势能面上各构型的能量,并进行了零点振动能(ZPE)校正,绘制出6种反应路径的势能面图。计算出各迁移反应过程中反应通道的反应总能量变化(ΔE)、化学反应焓变(ΔH)、吉布斯自由能变化(ΔG)。并利用Eyring的过渡态理论(TST)公式计算各反应通道的反应速率常数k[19]、以及不同温度下主反应通道的反应速率常数k,具体计算公式为
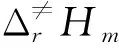
研究选用与参考文献 [20]相同的计算方法,对研究物质计算的结果与文献一致。
2 结果与讨论
2.1 稳定性分析
在B3LYP/6-31G(d,p)计算水平下PFHxA及其支链异构体的优化构型如图1所示,振动分析结果表示力常数矩阵本征值全为正,证明它们也是势能面上的稳定点。
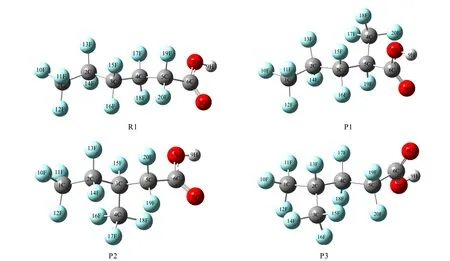
图1 全氟己酸(PFHxA)及其支链异构体的优化构型Fig.1 Optimized structures of PFHxA and its branched isomers
通过对气相和水溶液中PFHxA及其支链异构体的总能量计算可知,各物质在气相和水溶液中能量相差较小,其稳定性不受溶剂影响,可以通过理想气体状态预测水溶剂中PFHxA的迁移反应。
续图1 全氟己酸(PFHxA)及其支链异构体的优化构型Continued Fig.1 Optimized structures of PFHxA and its branched isomers
对于碳原子数目相同的三氟甲基单取代全氟戊酸(PFPeA),α位取代与β、γ位取代异构体相比较,α -三氟甲基异构体相对能量最低为-24.41 kJ/mol,更稳定。因为PFCAs是羧酸(RCOOH)R基中所有的H原子被F原子取代而生成的,F的原子半径以及电负性远大于H原子,所以导致C链结构发生扭曲,使得PFCAs具有螺旋结构。取代基(-CF3)的位置决定其空间位阻,从而影响到螺旋角度的改变。α位取代其螺旋角度改变较小,空间位阻最小,较为稳定。与能量分析一致,在单取代PFPeA中,α -三氟甲基全氟戊酸更为稳定。

表1 B3LYP/6-31G(d,p)计算水平下气相(g)、水溶液(l)中PFHxA及其支链异构体能量及螺旋扭曲角度Tab.1 The Energy and helix twist angle of of PFHxA and its branched isomers at the B3LYP/6-31G(d,p) level
2.2 反应机理
通过理论计算分析,在气相中,直链PFHxA可以迁移为α、β、γ位单取代的全氟戊酸,α位双取代、β位双取代的全氟丁酸,以及α、β位双取代的全氟丁酸。在B3LYP/6-31G(d,p)计算水平下,分别构建了直链PFHxA迁移转化为含有相同碳的支链PFCAs的反应途径:
(1) PFHxA→TS1→CF3CF2CF2CF(CF3)COOH (P1)
(2) PFHxA→TS2→CF3CF2CF(CF3)CF2COOH (P2)
(3) PFHxA→TS3→CF3CF(CF3)CF2CF2COOH (P3)
(4) PFHxA→TS1→IM1(P1)→TS4→CF3CF2C(CF3)2COOH (P4)
(5) PFHxA→TS1→IM1(P1)→TS5→CF3CF(CF3)CF(CF3) COOH (P5)
(6) PFHxA→TS3→IM2(P3)→TS6→CF3C(CF3)2CF2COOH (P6)
直链PFHxA反应过程中各驻点的零点能ZPE、总能量ET(含零点能校正)、相对能量ER和过渡态虚频(ν)见表2。从表2可知,过渡态均有且只有一个虚频,初步证明过渡态的正确性。根据过渡态内禀反应坐标(IRC)进行进一步计算,结果显示每一个过渡态振动分别指向各自对应的反应物和产物,证实他们是真实的过渡态。反应物、中间体和产物的振动分析结果表示力常数矩阵本征值全为正,证明他们也是势能面上的稳定点。
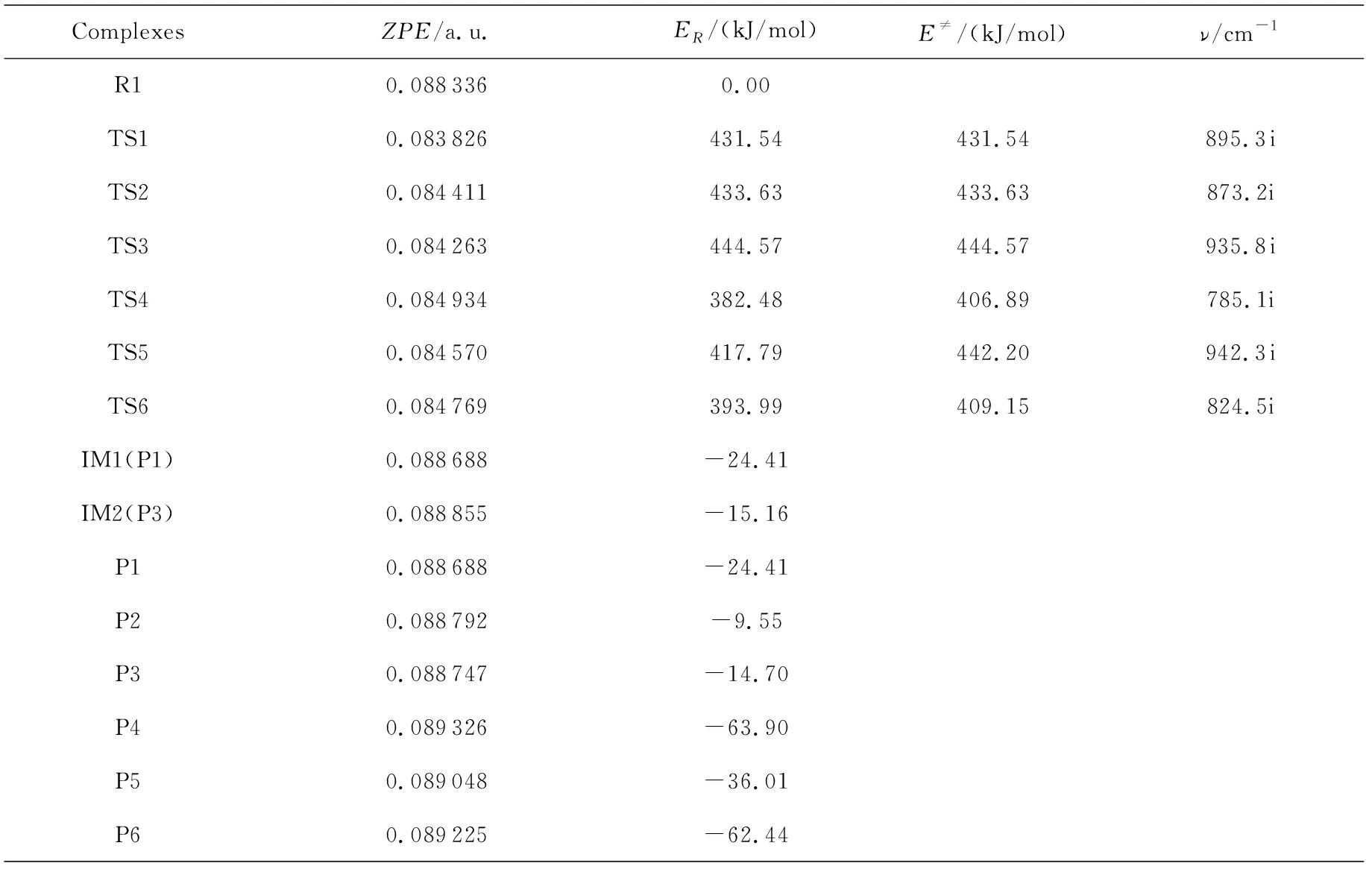
表2 B3LYP/6-31G(d,p)计算水平下直链PFHxA迁移反应中各驻点的能量及过渡态的虚频(ν)
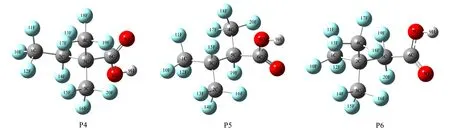
直链PFHxA反应过程中的过渡态、中间体、反应物和产物的优化分子构型及其结构参数如图2和图3所示。

图2 过渡态的优化构型Fig.2 Optimized structures of the transition states
2.2.1 单取代双三元环反应机理 从表2可知,反应物直链PFHxA分别吸收431.54 kJ/mol、433.63 kJ/mol、444.57 kJ/mol能量,经过双三元环过渡态TS1、TS2、TS3生成相对应的产物P1[CF3CF2CF2CF(CF3)COOH]、P2[CF3CF2CF(CF3)CF2COOH]、P3[CF3CF(CF3)CF2CF2COOH]。其螺旋角也发生改变,由初始的15.49°分别变为14.95°、7.80°、5.99°。
从图1至图3可以看出,通道(1)是由直链PFHxA迁移成α -三氟甲基全氟戊酸的反应,PFHxA吸收431.54 kJ/mol的能量生成过渡态TS1过程中,要完成F与C的迁移。首先,C(5)—F(20)键逐渐增长,由开始的0.135 0 nm增加到0.183 4 nm,键与键之间相互作用力减弱,有断键的趋势。相反,C(4)—F(20)之间开始成键,F(20)由C(5)迁移到邻位C(4),生成支链三氟甲基(-CF3)。与此同时,C(4)从主链开始迁移,∠C(3)-C(4)-C(5)由原来的114.3°减小为67.2°,C(4)变为支链C,C(3)-C(4)键长由0.155 7 nm增加到0.238 4 nm,逐渐开始断裂。C(3)-C(5)之间开始成键,键长为0.224 8 nm。C(4)-C(5)距离逐渐减小,从0.155 5 nm减少到0.137 9 nm。此时C(3)、C(4)、C(5)、F(20)形成双三元环结构,生成过渡态TS1。此反应过程为基元反应,经过过渡态TS1,直接反应得到最终产物P1[CF3CF2CF2CF(CF3)COOH]。同时在有水溶剂的情况下,对反应路径(1)的迁移过程进行分析。直链PFHxA可以通过相同的双三元环反应机理,吸收411.75 kJ/mol的能量,经过过渡态TS1,生成相同的产物P1[CF3CF2CF2CF(CF3)COOH]。

图3 中间体的优化构型Fig.3 Optimized structures of the intermediates
通道(2)是由直链PFHxA迁移成β-三氟甲基全氟戊酸的反应,PFHxA吸收433.63 kJ/mol的能量,通过键与键之间的断裂以及合成,形成双三元环过渡态TS2。TS2释放443.18 kJ/mol的热量,生成产物P2[CF3CF2CF(CF3)CF2COOH]。同样,通道(3)是由直链PFHxA迁移成γ-单取代的支链全氟羧酸的反应,直链PFHxA吸收了444.57 kJ/mol的能量,形成双三元环过渡态TS3,TS3释放449.27 kJ/mol的能量,反应生成产物P3[CF3CF(CF3)CF2CF2COOH]。
2.2.2 双取代的双三元环反应机理 从图2至图4(a)可以看出,反应物PFHxA迁移为双取代的全氟丁酸是非基元反应,需要经过两个过渡态和一个中间体,生成最终的产物α,α -双取代、β,β-双取代、α,β-双取代的全氟丁酸。
通道(4)是由直链PFHxA迁移成α位双取代全氟丁酸的反应路径,是在反应通道(1),生成IM1(α -单取代)的基础上,进行第二个支链的生成反应。中间体IM1跨越406.89 kJ/mol的能垒发生反应,生成过渡态TS4,在此过程中,C(3)与C(5)相连的F(20)靠近成键,键长为0.206 0 nm,C(5)-F(20)键长逐渐被拉伸,呈断键的趋势。在F进行迁移的同时,支链C也开始逐渐形成,C(3)由主链逐渐迁移为支链C,C(2)-C(3)键长增加到0.240 8 nm,有断裂的趋势,但C(3)与C(5)的键长开始缩短为0.139 9 nm,C(2)与C(5)开始成键。C(2)、C(3)、C(5)、F(20)逐渐开始形成双三元环结构,生成过渡态TS4,通过过渡态TS4,生成最终产物P4[CF3CF2C(CF3)2COOH]。
通道(5)是由直链PFHxA迁移成α、β位双取代全氟丁酸的反应路径。在反应通道(1),生成IM1(α -单取代)的基础上,进行第二个支链的生成反应。中间体IM1跨越442.20 kJ/mol的势垒发生反应,生成过渡态TS5,此过程中,C(2)与C(3)相连的F(16)靠近成键,C(3)-F(16)有拉伸断裂的趋势。C(1)与 C(3)有靠近成键的趋势,C(1)-C(2)有拉伸断裂的趋势。通过过渡态TS5,生成最终产物P5[CF3CF(CF3)CF(CF3)COOH]。
通道(6)是由直链PFHxA迁移为β位双取代全氟丁酸。在反应通道(3),生成IM2(γ-单取代)的基础上,进行第二个支链的生成反应。中间体IM2跨越409.15 kJ/mol的势垒发生反应,生成过渡态TS6,此过程中,C(4)与C(2)相连的F(13)靠近成键,C(2)-F(13)、C(4)-C(5),键长增加,有拉伸断裂的趋势。C(2)与C(5)之间开始成键。通过过渡态TS6,生成最终产物P6[CF3C(CF3)2CF2COOH]。
2.3 反应路径各物质NBO电荷分析
为了更进一步从电荷方面分析解释迁移反应机理,以反应路径1为例,研究了主反应过程中各驻点的电荷变化,用Gaussian NBO Version 3.1对主反应路径中各物质进行NBO电荷分析(表3)。

表3 PFHxA降解反应中速控步R1→P1各驻点物电荷分布(e)
从电荷方面分析,在R1经过过渡态TS1过程中,C(4)的正电荷由0.679e增加到0.957e,失去0.278e,同时C(3)、C(5)的正电荷逐渐减小,证明C(4)的电子向C(3)、C(5)转移,C(3)、C(5)电荷密度增加,得到电子,导致其活化,有利于C(3)-C(5)成键。与C(5)相连的F(20)接受周围F提供的电子,向C(4)靠近成键。在整个反应过程中,通过电子转移,实现C(4)-C(5)、C(5)-F(20)键断裂,C(3)-C(5)、C(4)-F(20)键形成。最终生成的产物为CF3CF2CF2CF(CF3)COOH。
2.4 能量分析
以相对能量作图所得直链PFHxA迁移为支链全氟羧酸的六条反应路径的总势能面图如图4(a)和图4(b),这六条反应路径同样互为竞争关系。路径(1)、(2)、(3)均为基元反应,可直接通过其反应势垒判断反应的难易程度。路径(4)、(5)、(6)为非基元反应,其反应难易程度由其决速步控制,各反应通道的决速步分别为R→IM1、R→IM1、R→IM2。通过比较各个反应路径的势垒,竞争过程通道(1)占优势,其反应势垒为431.54 kJ/mol,是主反应路径。
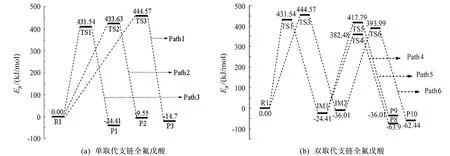
图4 直链PFHxA迁移为单取代支链和双取代支链全氟戊酸的反应路径势能面图Fig.4 Potential energy surface diagram of the reaction path of linear PFHxA migration to doublesubstituted branched chain and monosubstituted branched perfluorobutyric acid
由能量势垒图可知,直链PFHxA以迁移为α单取代全氟羧酸反应为主反应路径,但不论是主反应路径还是其他的反应路径,在298.15 K时,都需要跨越较高的势垒,需要向外界吸收大量的能量,反应较难进行,常温下很难实现由直链向支链的转化。
2.5 反应动力学分析
通过能量分析可知,迁移反应较难发生。为了进一步分析PFHxA迁移反应的难易程度。从动力学的角度,并利用Eyring的过渡态理论(TST)公式计算各反应通道的反应速率常数k[20]以及不同温度下主反应通道的反应速率常数k,见表4和表5。

表4 各反应通道相对于反应物的反应自由能、热力学性质(kJ/mol)及反应速率常数k(s-1)
由表4的各路径反应速率常数可知,PFHxA在迁移反应过程中,各反应路径的反应速率常数都很小,与能量分析一致,各个反应路径在常温下都较难实现。对于直链PFHxA,反应路径(4)、(5)、(6)分别为其对应的迁移为双取代PFCAs的非基元反应,其反应速率常数取决于决速步的速率常数,分别为1.73×10-60/s、1.73×10-60/s、5.37×10-65/s。与能量分析结果一致,六条反应路径中,反应路径(1)为PFHxA的主反应路径,其反应速率常数最大,反应较容易发生。
但即使是主反应路径,在常温下反应速率常数均达到10-60/s左右,反应亦难进行,为了探究温度对全氟羧酸迁移反应的影响,在B3LYP/6-31G(d,p)理论水平上分别计算300~1 000 K温度下PFHxA迁移主反应路径反应速率常数。
由表5可知,在300~1 000 K温度范围内,ΔG大于零,该反应在此温度范围内是非自发反应,反应不易发生。但随温度的升高,反应的速率常数k的数值明显增大,300~1 000 K范围内,反应速率常数提高了52个数量级。反应速率常数与温度呈正相关性。证明高温可以提高迁移反应的反应速率,更有利于实现直链到支链的迁移反应。但在自然界的常温条件下此迁移反应还是较难发生。
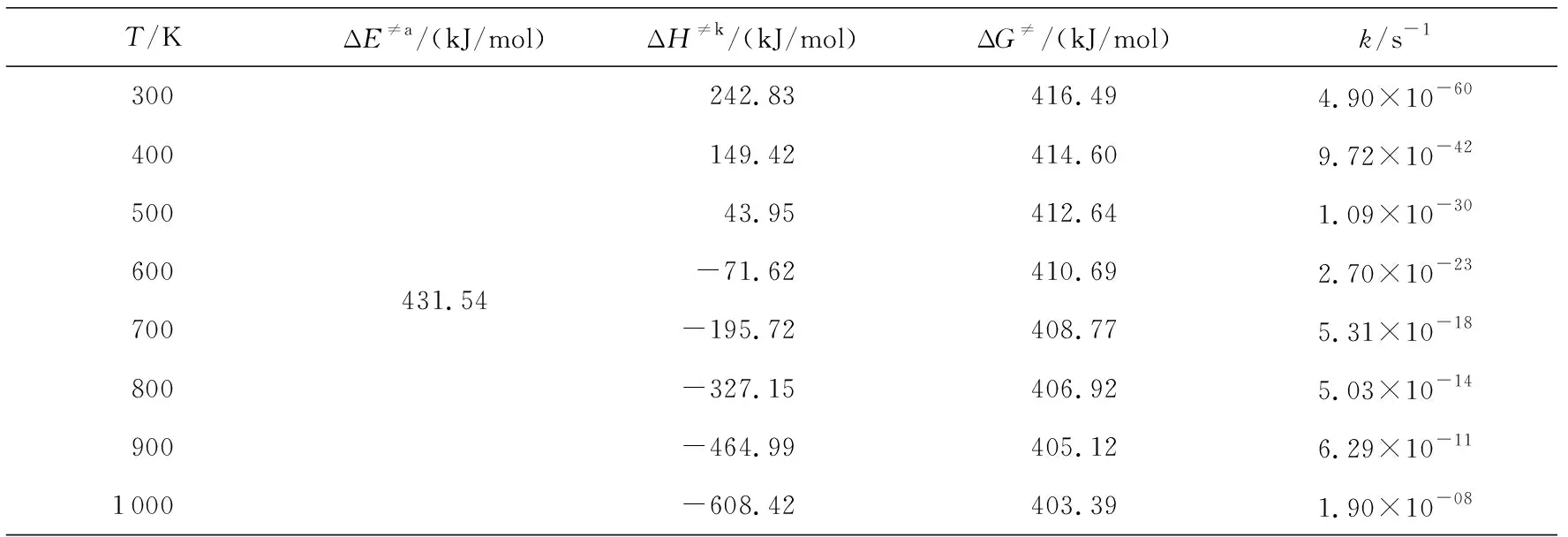
表5 300~1 000 K温度下主反应通道的热力学数据及反应速率常数Tab.5 Thermodynamic data and reaction rate constant of the main reaction channel at 300-1 000 K
3 结论
以PFHxA为例的直链PFCAs可迁移为相对应的支链PFCAs。根据能量分析,通过双三元环反应机理迁移为α -三氟甲基全氟戊酸的反应,相对其他反应路径较容易实现,是主反应路径,但在此过程中仍需要跨越较高的能量势垒。通过动力学分析,直链PFCAs的迁移反应是非自发反应,即使是主反应通道,其反应速率常数也很小。不论是从反应势垒还是反应速率常数分析考虑,在常温下,迁移反应均难进行。所以即使支链全氟羧酸比直链全氟羧酸更稳定,但在常温条件下,直链向支链迁移的过程中,因为需要跨越较高的能量势垒,向外界吸收大量的能量,所以反应较难发生。为了确保数据的准确性,对短链的全氟丁酸、全氟戊酸迁移反应进行计算分析。结果显示,与全氟己酸相同,常温下,迁移反应均难发生。研究初步从理论上对自然界中直链多于支链的现象进行了解释。但随着温度的升高,主反应通道的反应速率常数增加,迁移反应速率与温度呈正相关,高温有利于反应的进行。本研究从理论方面解释了自然界存在的问题,为后续的实验研究提供理论依据。
猜你喜欢
北京大学学报(自然科学版)(2022年3期)2022-06-17
包装工程(2022年1期)2022-01-26
农业机械学报(2021年10期)2021-11-09
中国农业科学(2021年7期)2021-04-21
农产品加工(2020年3期)2020-03-11
华声文萃(2019年12期)2019-09-10
北京航空航天大学学报(2019年3期)2019-04-08
分析化学(2019年3期)2019-03-30
分析化学(2015年7期)2015-07-30
安徽农业科学(2015年9期)2015-01-12
