
饵料蛋白水平对德国小蠊肠道细菌群落的影响
2021-08-07 02:11:46王亚茹田宝玉张碧尧范競文王国红
生态学报 2021年13期
王亚茹, 田宝玉, 张碧尧, 范競文, 戈 峰, 王国红,*
1 福建师范大学生命科学学院,细胞逆境响应与代谢调控福建省高等学校重点实验室, 福州 350108 2中国科学院动物研究所,农业虫害鼠害综合治理研究国家重点实验室, 北京 100101
昆虫肠道中定殖着大量的微生物,在昆虫食物消化和营养代谢中发挥着重要的功能,是调控宿主生物学性状和生态适应性的重要因素[1- 2]。大量的研究结果表明,昆虫肠道微生物数量和组成与宿主的食物密切相关。一方面,肠道微生物是宿主食物分解和消化的主要参与者,为宿主提供碳水化合物、维生素、氨基酸等营养和能量;另一方面,食物的结构和化学组成又影响肠道微生物的群落结构和功能[2]。采用桑叶饲养的家蚕(Bombyxmori),其肠道微生物种群比较一致,而采用不同人工饲料饲养的家蚕的肠道优势菌群有差异;相比于人工饲料,桑叶饲养的家蚕肠道微生物菌群更为丰富[3]。取食人工饲料和不同植物的小菜蛾(Plutellaxylostella)肠道细菌群落存在显著差异,小菜蛾肠道菌群在群落结构和代谢功能两方面均能协助宿主昆虫适应不同的食物[4]。因此,食物是决定昆虫肠道微生物种类和组成的重要因素之一[4- 6]。
蟑螂是杂食性昆虫,不同种类的蟑螂食性有一定的差异,食物的成分和含量对蟑螂的引诱、肠道菌群、营养和代谢产生影响[2,5,7- 8]。在发展蟑螂饵剂的研究中发现:饵料的营养组分是影响蟑螂取食量的重要因素, 蟑螂优先选择蛋白含量较高的饵剂[9]。本文前期研究也表明:饵料的蛋白含量影响德国小蠊雄成虫氮素营养利用效率。取食含25%—45%蛋白的饵料时,有利于雄成虫氮素营养利用;而高蛋白含量(65%)饵料增加德国小蠊代谢负担,不利于对食物的利用[7]。饵料蛋白水平过高也导致德国小蠊肠道菌群多样性、交配成功率和繁殖能力的下降[6, 8]。Yun 等发现野生德国小蠊与实验室喂养的德国小蠊相比,其肠道细菌具有更丰富的多样性,原因可能在于野生蟑螂食物高度多样化,多样化的食物能够提供蟑螂肠道细菌代谢所需的不同底物[10]。众所周知,蛋白质和氨基酸是昆虫食物的主要氮素营养,是影响宿主肠道微生物的主要因素[11- 12]。利用人工饲料饲养美洲大蠊(Periplanetaamericana)有利于拟杆菌门(Bacteroidetes)和厚壁菌门(Firmicutes)细菌的生长;不同饲料组成可以调节肠道细菌的群落组成和物种多样性;美洲大蠊取食高纤维食料时,厚壁菌门细菌数量显著增加;而取食高蛋白食料时,则促进其他细菌种群数量的增加[13-14]。利用不同蛋白含量的饵料喂养德国小蠊(Blattellagermanica)的雌虫,Pérez-Cobas 等发现菌群组成与特定的食物有关,肠道菌群在蟑螂氮源营养代谢中发挥重要的作用[6]。已有研究发现:昆虫不同虫态和性别间肠道菌群有差异[15-16]。如海灰翅夜蛾(Spodopteralittoralis)雌性成虫肠道以肠球菌属(Enterococcus)、克雷伯氏杆菌属(Klebsiella)和泛菌属(Pantoea)为主;雄性成虫则不同,其肠道菌群几乎全部由克雷伯氏杆菌属构成[15]。但是,蟑螂食性复杂,导致其肠道菌群的可塑性强,食物对蟑螂肠道菌群的调控能力和机制还不清楚[17]。不同蛋白水平饵料的影响在德国小蠊雄、雌虫之间是否有差异,以及影响的主要菌群及其代谢功能,目前并不清楚。
昆虫的肠道环境特殊,微生物种群复杂,其中含有大量的目前难以培养的微生物。因此,传统的体外纯培养技术严重低估了肠道微生物的多样性[18]。基于此,目前有关昆虫肠道微生物的研究,大都采用基于高通量测序的技术手段,可以更详尽深入地分析肠道微生物的种类、群落组成及变化[1- 2]。本研究采用高通量测序技术分析德国小蠊肠道细菌16S rRNA基因的V4区,鉴定德国小蠊雄成虫肠道菌群的多样性,评估不同蛋白质水平饵料对德国小蠊雄成虫肠道细菌群落结构和功能的影响,进一步探讨德国小蠊雄成虫肠道菌群功能,为蟑螂的诱捕和生物防治提供新思路。
1 材料与方法
1.1 试虫饲养
德国小蠊由广东省疾病预防控制中心赠送。蟑螂的饲养参照张碧尧等[7]和Wang等[19]的方法进行。
1.2 饵料配置
蟑螂饵料的配制在Hamilton等[20]和张碧尧等[7]方法的基础上加以修改。可消化的碳水化合物为糊精,将除含蛋白质成分外的所有干燥成分与 200 mL蒸馏水混合加热直至沸腾。待冷却至 50℃后加入酪蛋白和酵母(防止蛋白质变性),同时连续搅拌以防止凝结。将饵料倒入培养皿中,室温冷却数小时,在50℃下将饵料干燥一周,储存在干燥器中。最终制成蛋白质含量分别为5%和65%的人工饵料。蟑螂种群繁殖饵料为鼠粮(粗蛋白含量为25%),购自江苏省协同医药生物工程有限责任公司。
1.3 德国小蠊雄成虫饲养
将刚羽化的德国小蠊雄成虫单独饲养试验盒中(直径50 mm,高250 mm)使用鼠粮25%蛋白(CD1)、5%蛋白(LP2)、65%蛋白(HP3)等不同的饵料连续饲喂 21 d后,饥饿 24 h以去除非常驻细菌[11, 21]。每个处理重复3次。
1.4 德国小蠊肠道基因组DNA的提取及检测
取饥饿雄虫用75%的酒精对虫体表面消毒3次,每个重复3次,每次3 min,再用无菌生理盐水清洗5遍。在无菌生理盐水中解剖虫体,仔细分离出肠道,置于一个已灭菌的1.5 ml的离心管中,保存于-80℃冰箱中待用。采用 CTAB方法提取样本的基因组 DNA,利用琼脂糖凝胶电泳检测DNA的大小和完整度,并保存于-80℃冰箱中用于下一步高通量测序[6]。
1.5 德国小蠊肠道细菌16S rDNA V4区的高通量测序
以提取的德国小蠊肠道基因组总DNA为模板,选择融合测序引物和barcode序列的细菌通用引物(515F:5′-GTGCCAGCMGCCGCGGTAA- 3′;806R: 5′-GGACTACHVGGGTWTC TAAT- 3′)扩增细菌16S rDNA V4可变区。利用New England Biolabs 公司的 Phusion® High-Fidelity PCR Master Mix with GC Buffer和高效高保真酶进行PCR,确保扩增效率和准确性。PCR反应体系(30 μL):Phusion Master Mix(2 x) 15 μL,正向引物1.5 μL,反向引物1.5 μL,基因组DNA(1 ng/μL)10 μL,H2O 2 μL。反应程序:98℃预变性1 min;30个循环包括(98℃,10 sec;50℃,30 sec;72℃,30 sec);72℃,5 min。根据浓度将不同样品PCR扩增产物进行等摩尔浓度混样,充分混匀后使用1xTAE浓度2%的琼脂糖胶电泳纯化PCR产物,选择主带大小在400—450 bp之间的序列,割胶回收目标条带。使用Thermofisher公司的lon Plus Fragment Library Kit 48 rxns 建库试剂盒进行文库的构建,构建好的文库经过Qubit 定量和文库检测合格后,使用Thermofisher的lon S5TMXL进行上机测序。
1.6 生物信息学分析
1.6.1测序数据分析
使用Cutadapt(V1.9.1)[22]先对reads进行低质量部分剪切,再根据Barcode从得到的reads中拆分出各样品数据,截去Barcode和引物序列,初步质控得到原始数据(Raw reads) 经过以上处理后得到的Reads需要进行去除嵌合体序列的处理,Reads序列通过vsearch(https://github.com/torognes/vsearch/)与物种注释数据库进行比对检测嵌合体序列,并最终去除其中的嵌合体序列,得到最终的有效数据(Clean Reads)。
1.6.2OTU聚类和物种注释
利用Uparse软件(v7.0.1001)[23]对所有样品的全部 Clean Reads 进行聚类,默认以97%的一致性将序列聚类成为OTUs(Operational Taxonomic Units),同时选取OTUs的代表性序列,依据其算法原则,筛选OTUs中出现频数最高的序列作为OTUs的代表序列。 对OTUs序列进行物种注释,用Mothur方法[24]与SILVA132的SSU rRNA数据库[25]进行物种注释分析(设定阈值为0.8—1),获得分类学信息并分别在各个分类水平:kingdom(界),phylum(门),class(纲),order(目),family(科),genus(属),species(种)统计各样本的群落组成。使用MUSCLE(Version 3.8.31)软件[23]进行快速多序列比对,得到所有OTUs序列的系统发生关系。最后对各样品的数据进行均一化处理,以样品中数据量最少的为标准进行均一化处理,后续的Alpha多样性分析和Beta多样性分析都是基于均一化处理后的数据。
1.6.3德国小蠊肠道细菌多样性分析
基于得到的OTU表,使用VennDiagram包绘制Venn图揭示样品间共有的OTUs。使用Qiime软件包(Version 1.9.1)计算各个样品的Alpha多样性并绘制OTU稀释曲线。Alpha多样性用来衡量样品内的物种多样性和丰度差异,主要通过Observed-OTUs,Chao1,Shannon,Simpson,ACE,Goods-coverage,PD-whole-tree等指标来表达。使用R软件(Version 2.15.3)进行Alpha多样性指数组间差异分析。采用QIIME进行基于unifrac距离矩阵的β-多样性并绘制NMDS图来评估样本间的相似度及不同样本群落组成相似性和差异。LEfSe分析使用LEfSe软件,默认设置LDA Score的筛选值为4。
1.6.4功能注释
基于微生物群落组成的功能预测参照Kakumanu等[26]和王天召等[27]的方法。测序样品以SILVA数据库序列为参考序列聚类出OTU,进而获取功能注释信息。利用BLASTN算法将其比对到SILVA SSU Ref NR 数据库(BLAST bitscore >1500)建立相关矩阵,将通过UProC和PAUDA两种方法注释的KEGG数据库原核生物全基因组功能信息对应到SILVA数据库中,实现SILVA数据库功能注释。
2 结果
2.1 德国小蠊肠道细菌16S rDNA测序结果处理和统计
OTU是在系统发生学或群体遗传学的研究中,给某一个分类单元(属、种和分组等)设置的一个标志,通常相似度为97%以上可以分为一个 OTU。对不同蛋白水平处理组德国小蠊肠道细菌有效测序数据进行统计,总测序标签为724954条,平均标签数80550.4条,测序数目较大,可以充分保证数据的准确性。其中CD1.2的总标签最多,为84195条(表1)。采用对测序序列进行随机抽样,以抽到的序列数与它们所代表的OTU数目构建德国小蠊肠道细菌高通量测序结果的稀释性曲线(图1)。从图1可以看出物种稀释曲线趋向于平行,表明目前获得的测序数据量合理,能够反映德国小蠊肠道细菌的多样性。
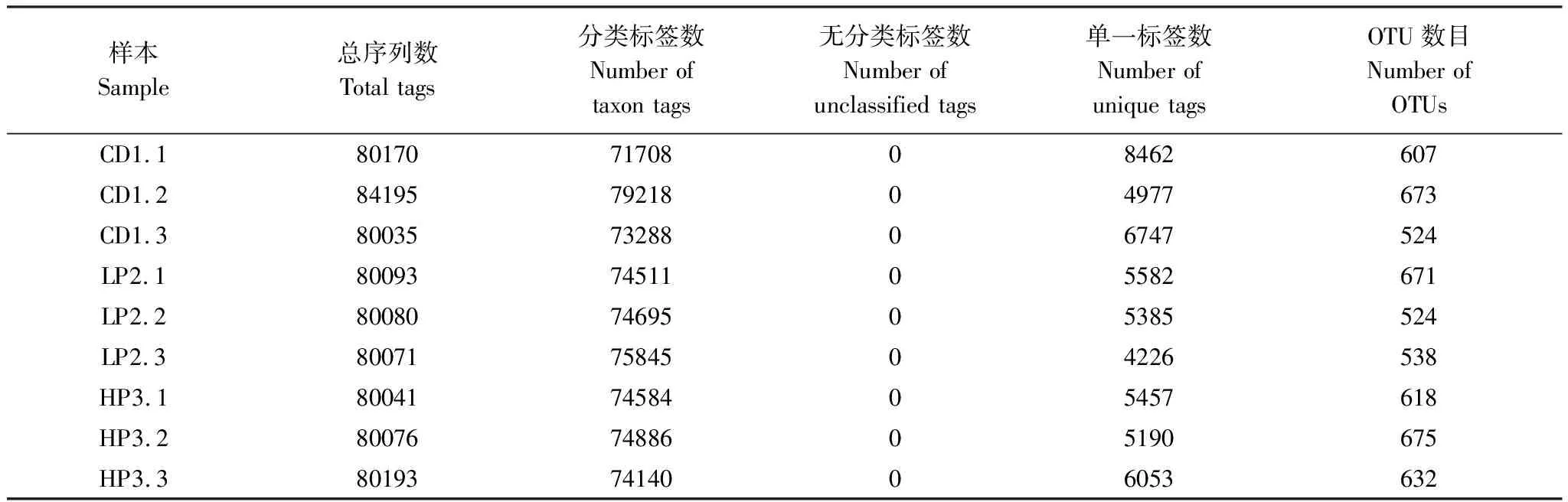
表1 德国小蠊肠道细菌16S rDNA测序有效数据统计
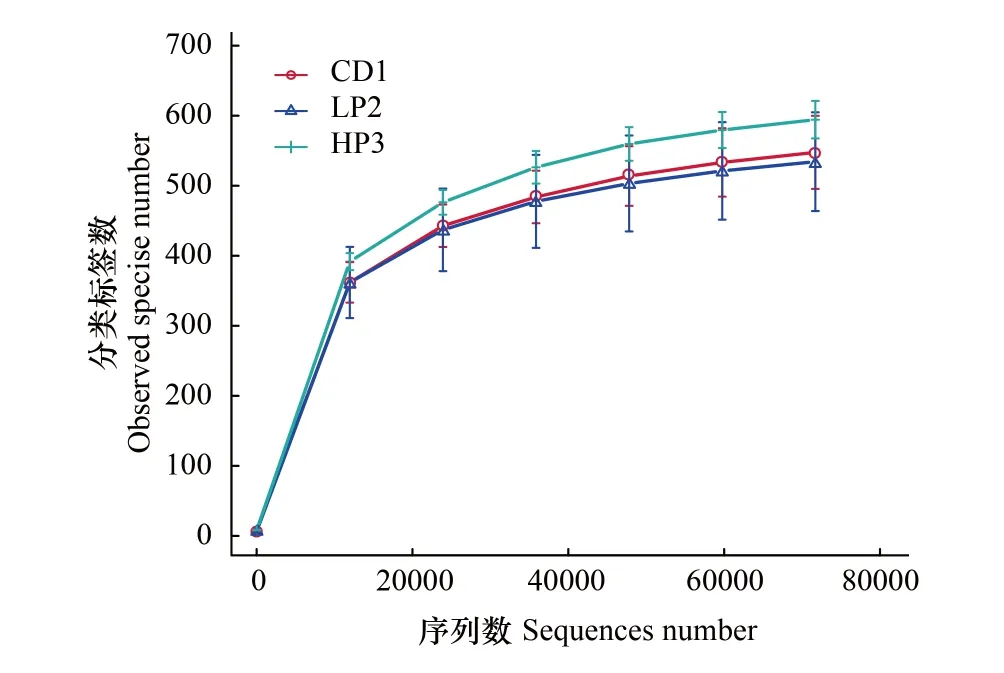
图1 德国小蠊肠道细菌物种稀释曲线 Fig.1 Observed species rarefaction curves of the intestinal bacteria in B. germanicaCD1: 鼠粮饲喂德国小蠊组; LP2: 5%蛋白饵料饲养组; HP3: 65%蛋白饵料饲养组
2.2 德国小蠊肠道细菌的群落组成和丰度
基于OTUs注释结果,所测样本在门水平分布最多的优势菌为拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)、梭杆菌门(Fusobacteria)和厚壁菌门(Firmicutes)。CD1组的优势菌门为拟杆菌门、变形菌门和厚壁菌门,相对丰度依次为46.61%、38.33%和8.43%。LP2组的优势菌门为拟杆菌门、变形菌门和厚壁菌门,相对丰度依次为71.39%、10.17%和13%。HP3组优势菌门为拟杆菌门、变形菌门、梭杆菌门和厚壁菌门,相对丰度依次为50.56%、18.4%、15.6%和9.75%。其中CD1组脱铁杆菌门(Deferribacteres)丰度显著低于LP2(df1,2=1,4,F=16.394,P<0.05),HP3组梭杆菌门丰度显著高于其他两组(df1,2=2,6,F=29.238,P<0.05)(图2)。
在科的水平上,德国小蠊肠道菌群共检测到可鉴定的细菌10个科。这些类群的细菌在3个处理的样本中均有出现。CD1组相对丰度大于5%的优势菌科分别是黄单胞菌科(Xanthomonadaceae)、(unidentified Bacteroidales)、拟杆菌科(Bacteroidaceae)、脱硫弧菌科(Desulfovibrionaceae)和(Tannerellaceae),相对丰度依次为27.94%、16.02%、7.04%、6.97%和6.71%。LP2组相对丰度大于5%的优势菌科是拟杆菌科、理研菌科(Rikenellaceae)和(Tannerellaceae),相对丰度依次为47.44%、5.87%和5.84%。HP3组相对丰度大于5%的优势菌科是拟杆菌科、黄单胞菌科、梭杆菌科(Fusobacteriaceae)和(unidentified Bacteroidales),相对丰度依次为23.97%、15.55%、6.44%和6.19%。其中CD1组拟杆菌科丰度显著低于HP3组(df1,2=1,4,F=32.996,P<0.05),HP3组梭杆菌科丰度显著高于其他两组(df1,2=2,6,F=31.618,P<0.05)(图3)。
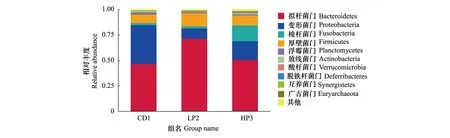
图 2 德国小蠊肠道细菌门水平上的物种相对丰度Fig.2 Histogram of relative abundance of the intestinal bacteria in B. germanica at the phylum level
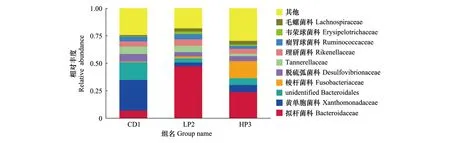
图3 德国小蠊肠道细菌科水平上的物种相对丰度Fig.3 Histogram of relative abundance of the intestinal bacteria in B. germanica at the family level
在属的水平上,德国小蠊肠道菌群主要由拟杆菌属(Bacteroides) 、寡养单胞菌属(Stenotrophomonas)、梭杆菌属(Fusobacterium)、营发酵单胞菌属(Dysgonomonas)、脱硫弧菌属(Desulfovibrio)、另枝菌属(Alistipes)、蟑螂杆状体属(Blattabacterium)等组成。CD1组相对丰度大于1%的优势菌属为寡养单胞菌属、营发酵单胞菌属、拟杆菌属、脱硫弧菌属、另枝菌属和塞巴鲁德氏菌属(Sebaldella),相对丰度依次为27.94%、11.15%、7.04%、6.85%、2.97%和1.21%。其他未鉴定的细菌占比38.97%。在LP2组中,拟杆菌属占绝对优势,其相对丰度为47.44%。相对丰度大于1%的还有另枝菌属、脱硫弧菌属、寡养单胞菌属、营发酵单胞菌属、(CandidatusSoleaferrea)、(Erysipelatoclostridium)和梭杆菌属,相对丰度分别为4.75%、3.74%、3.39%、3.33%、1.93%。1.91%和1.76%;未鉴定的细菌占比为29.19%。HP3组相对丰度大于1%的优势菌属为拟杆菌属、梭杆菌属、寡养单胞菌属、脱硫弧菌属、另枝菌属、营发酵单胞菌属、蟑螂杆状体属、(unidentifiedBurkholderiaceae),其相对丰度依次为23.97%、15.54%、6.43%、4.37%、3.12%、2.91%、2.17%和1.47%。其他未鉴定的细菌占比为32.98%。CD1组与LP2组相比,营发酵单胞菌属丰度显著高于后者(df1,2=1,4,F=10.137,P<0.05);CD1组拟杆菌属丰度显著低于HP3组(df1,2=1,4,F=32.996,P<0.05);HP3组梭杆菌属丰度极显著高于其他两组(df1,2=2,6,F=31.6,P<0.01)(图4)。
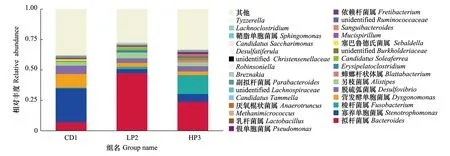
图4 德国小蠊肠道细菌属水平上的物种相对丰度Fig.4 Histogram of relative abundance of the intestinal bacteria in B. germanica at the genus level
OTUs分析表明CD1组有814个 OTUs,LP2组有785个OTUs, HP3组有881 OTUs。维恩图分析发现CD1组和LP2组共有657个的 OTUs,CD1组和 HP3组共有665个OTUs,LP2组和 HP3组共有655个OTUs,3组样本共有的 OTUs数目为585个,三个处理组的菌群具有很高的相似性,表明尽管不同的蛋白质饵料饲育对肠道菌群有明显影响,但主要菌群保持较高的稳定性(图5)。
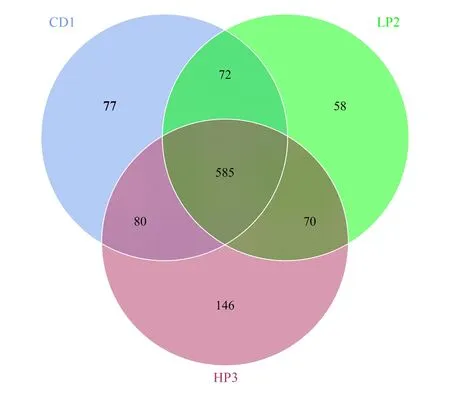
图5 德国小蠊肠道细菌venn图Fig.5 Venn diagram of the intestinal bacteria in B. germanica图中数字表示德国小蠊肠道细菌的数目
2.3 德国小蠊肠道细菌多样性特征
Alpha 多样性统计如表2,ACE指数和Chao1指数反映样品中群落的丰富度,数值越大,群落丰富度越高; Shannon指数越高和Simpson指数越低反映群落的多样性越高。该结果表明这三种饵料喂养德国小蠊肠道菌群具有高的物种多样性和丰富度,但三组饵料之间没有显著差异。
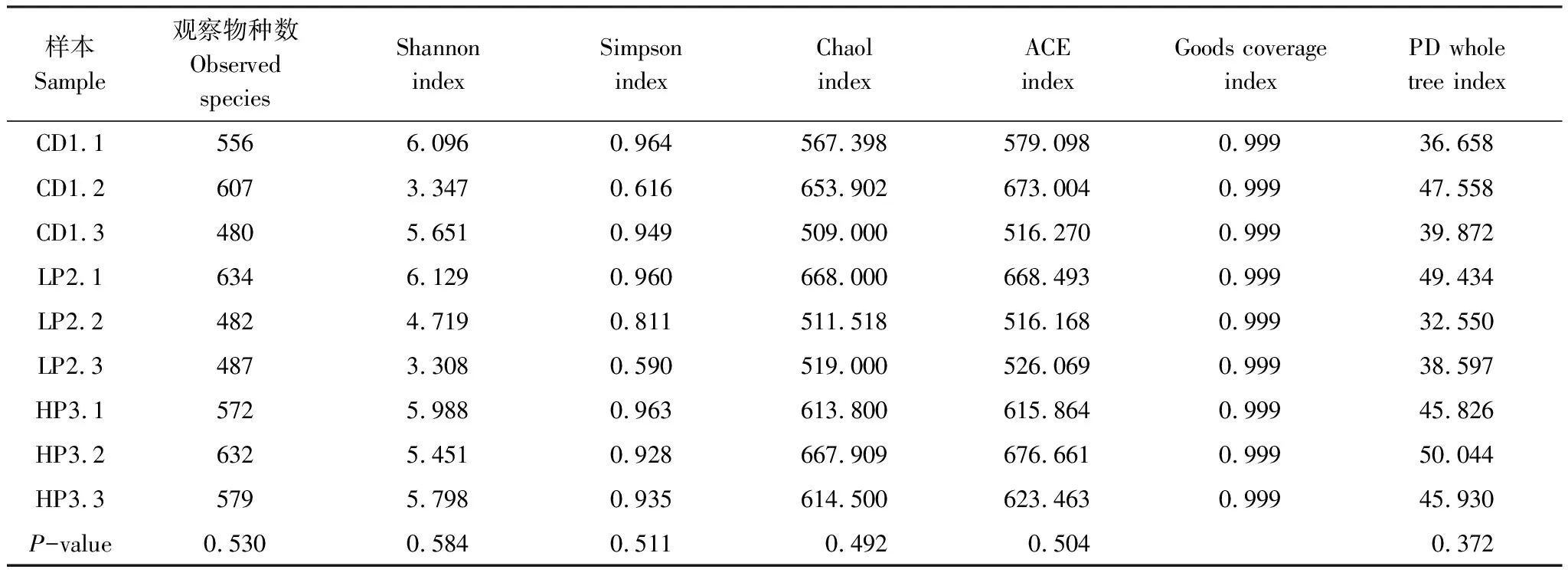
表2 德国小蠊肠道细菌α多样性指数
基于Bray-Curtis相似性系数的NMDS分析发现,3组处理的三个平行样本分布较为离散,说明3组样品组间物种群落相似性系数较低,但HP3组与另外两组的距离更远,表明高蛋白处理样本与低蛋白处理(小于25%)样本的肠道菌群组成具有较大的差异(图6)。
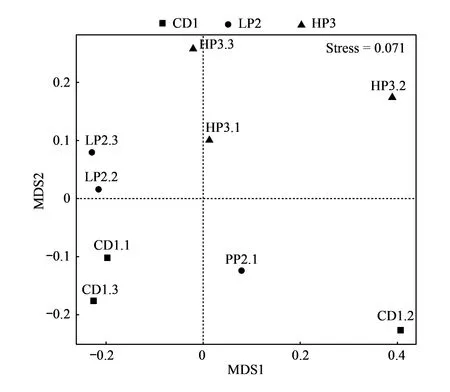
图6 德国小蠊肠道细菌的NMDS图Fig.6 NMDS diagram of the intestinal bacteria in B. germanicaNMDS: 非度量多维尺度分析Non-metric multidimensional scaling
2.4 德国小蠊肠道细菌组间物种差异
LDA值分布柱状图展示了德国小蠊雄成虫肠道细菌3组样本中在丰度上具有明显差异的物种。分析表明,在CD1组中丰度上有明显差异的物种为变形菌门(Proteobacteria)和包西氏菌属(Bosea)。LP2组在丰度上有明显差异的物种为(Lamiaceae)、(unidentified Acidimicrobiia)和(Lamia)。HP3组丰度上有明显差异的物种为梭杆菌科(Fusobacteriaceae)、梭杆菌属(Fusobacterium)、变异梭杆菌(Fusobacteriumvarium);伯克霍尔德科(Burkholderiaceae)以及(unidentified Gammaproteobacteria)(图7)。
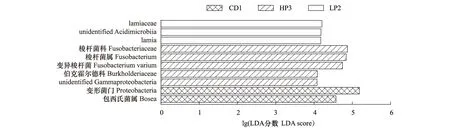
图7 LEfSe分析德国小蠊肠道细菌分类差异Fig.7 LEFSe analysis of the intestinal bacteria in B. germanica
2.5 德国小蠊肠道细菌的功能预测
运用Tax4Fun技术,在德国小蠊雄成虫肠道样品中共发现了44个基因簇。肠道细菌菌群功能基因和代谢途径分析表明,KEGG代谢通路中德国小蠊肠道细菌编码的大多数基因与其代谢功能有关。在发现的基因簇中,优势基因簇包括代谢(Metabolism)、遗传信息处理(Genetic Information processing)和环境信息处理(Environmental information processing)(图8)。选取丰度排名前35的功能及它们在每个样品中的丰度信息绘制热图,并从功能差异层面进行聚类(图9),HP3组肠道菌群能量代谢基因的相对丰度极显著高于CD1组(df1,2=1,4,F=102.86,P<0.01),外源性物质代谢与降解基因的相对丰度极显著高于LP2组(df1,2=1,4,F=19.973,P<0.01),其他氨基酸代谢基因的相对丰度显著高于LP2 组(df1,2=1,4,F=16.564,P<0.05)。
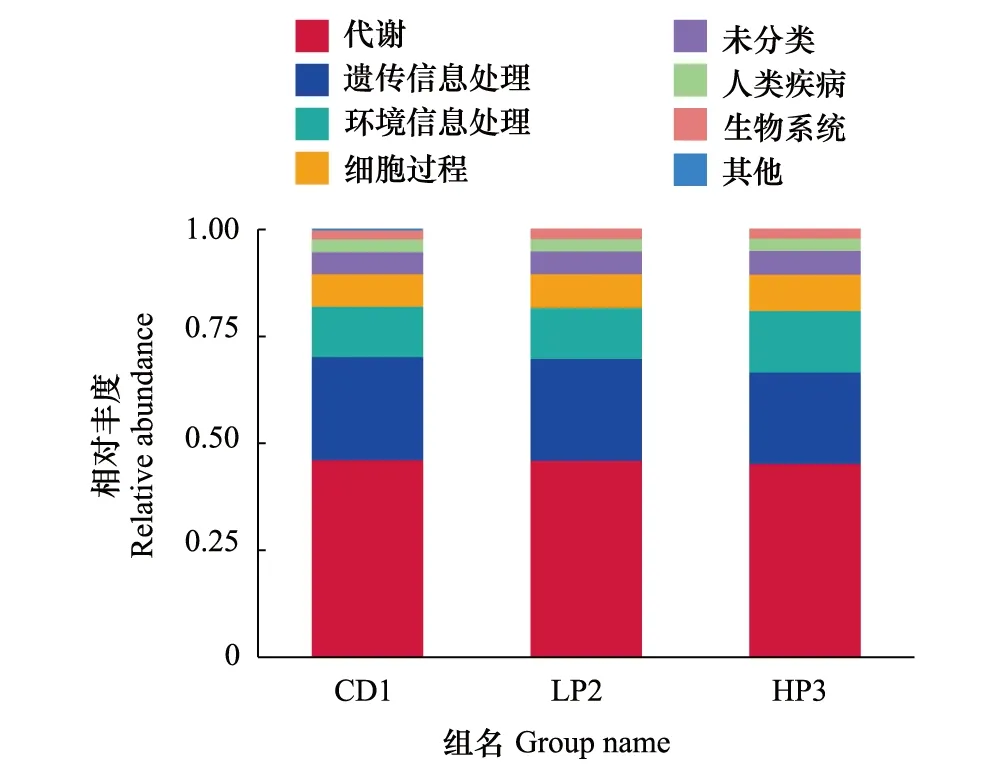
图8 KEGG通路Fig.8 KEGG pathway
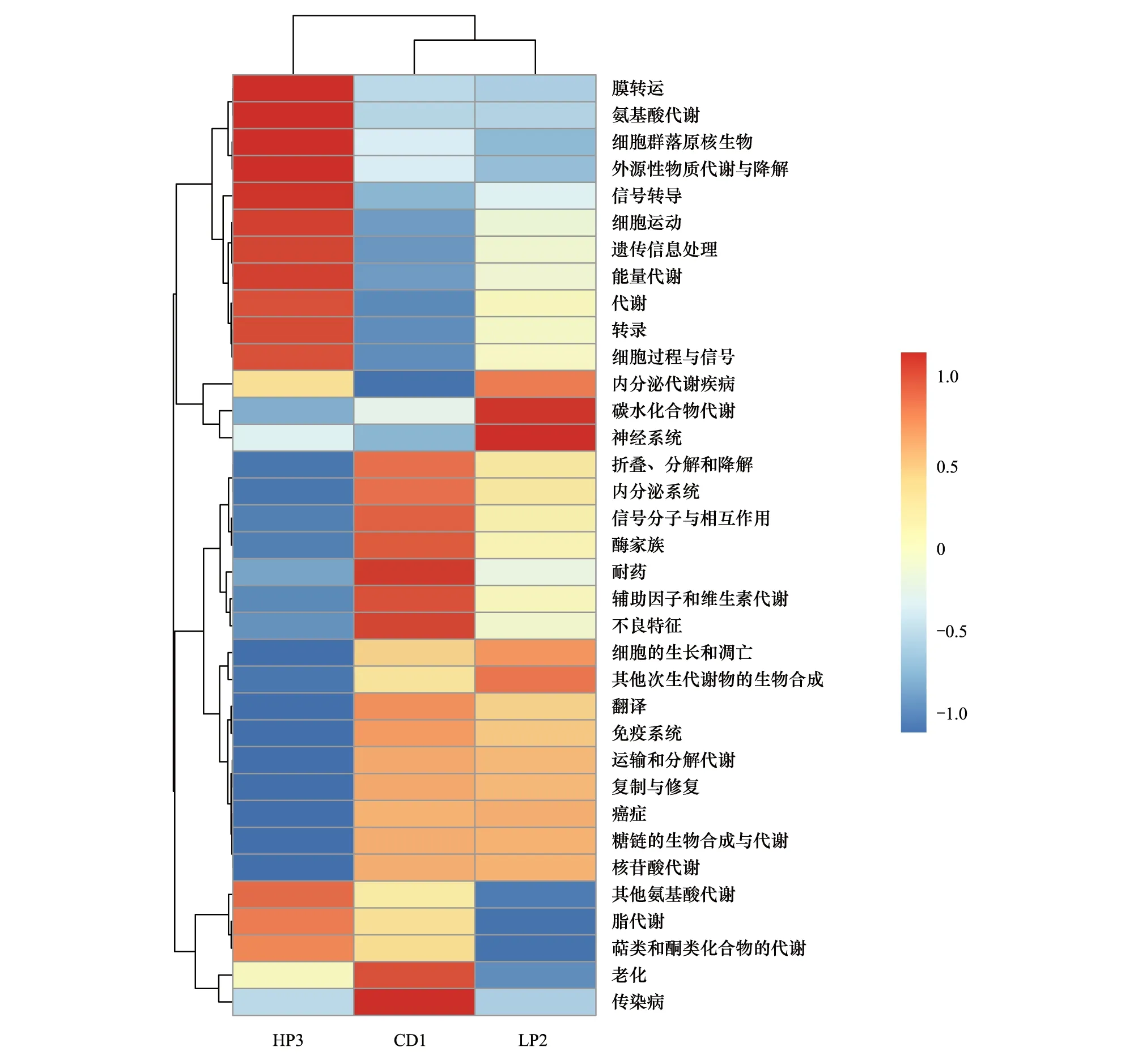
图9 Tax4Fun功能注释聚类热图Fig.9 Tax4Fun functional annotation clustering heat map
3 讨论
德国小蠊雄成虫肠道菌群门水平上优势菌为拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)、梭杆菌门(Fusobacteria)和厚壁菌门(Firmicutes);科水平上优势菌主要属于拟杆菌科、黄单胞菌科、梭杆菌科和脱硫弧菌科等。这与Pérez-Cobas等[6]、Kakumanu等[26]、Carrasco等[28]、Rosas等[29]和Chao等[30]对德国小蠊肠道菌群的研究结果相似,说明这些类群的细菌是德国小蠊肠道内的核心微生物(core microbes)。比较分析发现,德国小蠊雄成虫肠道优势菌群与褐飞虱(Nilaparvatalugens)[27]、滨海油葫芦(Teleogryllusoceanicus)[31]和印度谷蛾(Plodiainterpunctella)[32]肠道优势菌群相类似,一些昆虫的肠道微生物在高级分类单元上可能存在一定的相关性。
相对于低蛋白(5%)饵料,高蛋白饵料(65%)饲养的德国小蠊雄虫肠道细菌物种多样性没有显著差异,这一结果支持Pérez-Cobas等[6]对德国小蠊雌虫的研究结论。在德国小蠊研究中发现:与野生的蟑螂种群相比,实验室种群的肠道细菌多样性下降[6, 28];饲养时间增长有利于细菌物种多样性的增加[28]。野生蟑螂的肠道中含有数量众多的低丰度细菌种群,这些低丰度种群在实验室饲养过程中消失了[14, 33]。实验室饲养蟑螂肠道细菌多样性下降,可能与人工饵料成分比较固定、成分相对简单有关。饵料蛋白含量对昆虫肠道菌群的作用,在果蝇(Drosophilaspp)[34]、大头金蝇(Chrysomyamegacephala)[35]和印度谷蛾[32]等昆虫中也做过探讨;高蛋白饵料饲养降低果蝇、大头金蝇和印度谷蛾肠道菌群的物种多样性。由于不同研究所使用的饵料(包括蛋白质来源、种类和质量)不同,昆虫的饲养时间也不同,要作真正意义上有价值的比较,目前还存在困难。
不同蛋白质含量饵料饲养德国小蠊雄虫后,肠道细菌中不同种群的相对丰度发生显著的改变;高蛋白饵料饲养促进黄单胞菌科和梭杆菌科细菌富集,低蛋白饲料饲养则有利于拟杆菌科、理研菌科和(Tannerellaceae)细菌的生长。在美洲大蠊的研究中,Tinker和Ottesen没有作昆虫性别的区分,但发现不同饵料(包括高脂肪、高碳水化合物和高蛋白)对美洲大蠊肠道菌群的丰度没有显著的影响[14]。在德国小蠊雌虫肠道菌群中,相对丰度变化最大的是紫单胞菌科(Porphyromonadaceae)和梭杆菌科(Fusobacteriaceae);无蛋白饵料饲养促进紫单胞菌科细菌的生长,其相对丰度为37%,高于正常蛋白和高蛋白饲料组(分别为29%和26%);无蛋白饵料饲养不利于梭杆菌科细菌生长,其相对丰度为7%;显著低于正常蛋白和高蛋白饲料组(分别为32%和23%)[6]。此外,德国小蠊雌虫中没有发现黄单胞菌科的细菌[6],而在本研究中却发现黄单胞菌科细菌是德国小蠊雄成虫肠道的优势菌群。这种雌、雄虫之间细菌种群分布的差异是否与其性别分化有关,值得后续深入研究。
由不同蛋白水平饵料饲养而导致的德国小蠊雄虫肠道细菌种群相对丰度发生改变,可能与饵料成分对不同类群细菌的选择性相关。食物改变导致肠道细菌种群发生波动在德国小蠊和其他昆虫均有发现[6,8, 13- 15, 35],但食物结构和组分影响昆虫肠道菌群的具体机制目前不太清楚。在长期的进化过程中,不同的微生物类群适应各自的生长环境,进化出对不同有机质利用能力的差异[1, 5, 36]。野生蟑螂肠道微生物群落更为丰富,可能与野生蟑螂的食料更为多元化有关。野生蟑螂肠道中定殖着大量的低丰度细菌种群[33, 37],其中的变形菌门细菌大多数与生物固氮有关,在无氮或贫氮环境条件下,就需要这些菌群帮助宿主进行氮素的固定和循环[5, 11]。蛋白组分析结果显示:大多数拟杆菌科细菌含有较多的分解多糖的酶蛋白,主要功能可能是帮助宿主降解复杂的碳水化合物,产生简单而易于利用的糖类。因此,饵料中高比例的复杂碳水化合物有利于拟杆菌的生长[13, 28]。在人体肠道中,拟杆菌科细菌同样适应于高蛋白食物;拟杆菌分解复杂多糖和蛋白质后产生的单糖和氨基酸,则促进肠道中梭杆菌的积累[38]。因此推测,在本研究设计的饵料中,低蛋白饵料中高水平的糊精(85%)有利于拟杆菌的生长,因而在低蛋白饵料饲养蟑螂肠道中拟杆菌科细菌占绝对优势(47.44%);而在高蛋白饵料中,较高水平的糊精(13%)和高水平的蛋白(65%),则促进拟杆菌和梭菌的协同生长。菌群功能注释显示高蛋白组德国小蠊肠道里能量代谢、氨基酸代谢和物质分解相关的基因表达显著提高(图10),和梭杆菌属细菌丰度升高同步,推测梭杆菌属细菌可能与饵料中蛋白质降解和能量利用相关,缓解饵料中过多的蛋白质对德国小蠊的代谢负担。但高蛋白饵料对德国小蠊雄虫肠道细菌种群的调节作用由何种因素主导,以及梭杆菌属细菌的富集在高蛋白水平条件下的作用有待于深入的研究。
不同蛋白质水平饵料饲养的德国小蠊其肠道菌群结构发生显著的变化,这些变化可能与德国小蠊对食物的适应有关,提示肠道细菌在德国小蠊对环境和食物的生存适应上可能发挥着重要的作用。本文的研究结果有利于进一步将昆虫肠道菌群与昆虫的取食、代谢和发育联系起来,揭示相关的作用机制,从而为利用肠道菌发展饵料,实现对包括德国小蠊在内的卫生害虫治理奠定基础。
致谢:感谢福建农林大学植物保护学院夏晓峰副教授对文章写作的帮助。
猜你喜欢
青少年科技博览(中学版)(2022年9期)2022-11-01 08:21:46
领导文萃(2022年15期)2022-07-06 07:13:48
杂文选刊(2022年7期)2022-06-30 11:18:35
文萃报·周五版(2022年15期)2022-04-21 12:15:19
少年文艺·我爱写作文(2021年2期)2021-01-11 08:44:50
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
水生生物学报(2019年4期)2019-07-20 08:08:10
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
小猕猴学习画刊(2017年12期)2017-12-26 12:08:34
