
新生儿家族性非典型溶血性尿毒综合征1例基因分析并文献复习*
2021-08-06 08:52:32刘思英常立文蔡保欢
华中科技大学学报(医学版) 2021年4期
刘思英, 常立文, 蔡保欢, 王 靓, 谢 敏, 刘 伟
华中科技大学同济医学院附属同济医院新生儿科,武汉 430030
溶血性尿毒综合征(hemolytic uremic syndrome,HUS)是血栓性微血管病(thrombotic microangiopathy,TMA)的类型之一,表现为典型的微血管病溶血性贫血、血小板减少和肾功能衰竭三联征[1]。国际溶血性尿毒综合征组织将该病分类为:①感染性腹泻相关性HUS,病原为产生志贺毒素的大肠埃希菌、痢疾志贺菌、肺炎链球菌、H1N1病毒;②继发性HUS,继发于具有相似病理改变的一些共性疾病,比如造血干细胞或实体器官移植,恶性肿瘤,自身免疫性疾病,艾滋病,妊娠,溶血、肝酶升高及血小板减少综合征(HELLP综合征),系统性红斑狼疮,抗磷脂抗体综合征,恶性高血压,原发性肾病以及药物诱导(最常见的奎宁、环孢素和他克莫司)和使用避孕药;③先天性或获得性钴胺素C缺乏相关性HUS;④遗传性或获得性非典型溶血性尿毒综合征(aHUS),病理机制是补体因子及二酰基甘油激酶ε(diacylglycerol kinase epsilon,DGKE)基因突变导致补体替代途径失调。DGKE是一种含有花生四烯酸的二酰甘油信号的抑制剂[2],缺少该酶会导致一种血栓前状态。aHUS是HUS中一种少见的可威胁生命的补体失调性疾病,也会表现出经典的三联征。与典型HUS截然相反,aHUS预后差,病死率高,超过半数患者会发展为终末期肾病,发病年龄跨度大,从胎儿期到成年都有发病报道。约60%的aHUS是遗传性的,这类aHUS即使治疗完全恢复后也常常会复发[3]。aHUS的病因是补体调控因子蛋白H因子(complement factor H,CFH)、补体调控蛋白B因子(complement factor B,CFB)、补体调控蛋白I因子(complement factor I,CFI)、补体C3(C3)、膜辅助蛋白(membrane cofactor protein,MCP)及拮抗CFH的抗体(CFHR)的基因变异从而导致相应补体调节系统失调[3-4]。诊断aHUS需要首先确诊为TMA,在排除志贺毒素相关性HUS、血栓性血小板减少性紫癜(thrombotic thrombocytopemic purpura,TTP)及其他引起TMA的病因后,再对补体水平进行相对全面的评估,包括补体调节因子(CFH、CFHR1/3/4、CFHR、CFI和MCP)和补体活化因子(CFB和C3)在血清中的浓度以及相应基因检测。我们报道了一种aHUS易感性基因变异,基因检测表现为染色体1q31处补体激活基因簇调控因子中一个较大基因组片段(57 kb)的杂合缺失,涉及CFHR1和CFHR3两个蛋白,该变异导致发生新生儿非典型溶血性尿毒综合征。同时在患儿父亲相同基因外显子区域也找到大片段杂合缺失,验证了患儿为先天性遗传,致病基因来自父亲,出生即发病。父亲携带突变基因而未发病,与文献中认为补体基因突变赋予了疾病的遗传易感性,而不是直接致病的观点吻合,即突变基因不完全外显[5]。本文回顾性分析华中科技大学同济医学院附属同济医院诊治的1例新生儿aHUS的临床资料和家系基因变异特征,并结合文献进行综合分析。
1 临床资料
患儿,男,出生2 h,G3P2,双胎之一,35周+1,出生体重(BW)2480 g。因另一胎胎心消失,紧急剖宫产。剖宫后发现另一胎死亡,患儿生后5 min、10 min Apgar评分分别为8分、9分,羊水清,脐带、胎盘无异常。生后因发现患儿皮肤苍白,外院立即查血常规示血红蛋白80 g/L,血小板94×1012/L,无呻吟、吐沫。为求进一步诊疗,遂转至我院新生儿科。既往史:父母ABO血型均为O型,产前促胎肺成熟治疗2 d。否认家族遗传病史。
入院后辅助检查:入院当天血常规+网织红细胞计数:白细胞计数14.54×109/L,中性粒细胞(%)70.6%(上升),红细胞计数2.10×1012/L(下降),血红蛋白79.0 g/L(下降),血小板计数84.0×109/L(下降),网织红细胞(%)7.64%(上升),幼稚网织红细胞(%)59%(上升)。C反应蛋白(CRP)<0.5 mg/L,降钙素原(PCT)12.5 ng/mL。生化:谷丙转氨酶63 U/L(上升),谷草转氨酶303 U/L(下降),总胆红素29.4 μmol/L,间接胆红素19.5 μmol/L,血尿素氮(BUN)4.20 mmol/L,肌酐70 μmol/L,乳酸脱氢酶超过1867 U/L,碳酸氢根18.5 mmol/L(下降),肌酸激酶818 U/L(升高),钾4.99 mmol/L,钠139.5 mmol/L,氯106.9 mmol/L,钙1.97 mmol/L,磷1.93 mmol/L。直接抗人球蛋白试验(Coombs’试验)可疑阳性(±)。凝血功能正常。粪常规无异常。尿常规:红细胞3+,尿蛋白3+,红细胞计数超过20000/μL,白细胞计数47.70/μL(上升)。尿红细胞位相镜检异形RBC(%)20%(上升)。外周血涂片:成熟红细胞大小不均,可见少数靶型红细胞,同时可见少数红细胞碎片;血小板分布减少,形态大致正常。肾脏彩超:①左肾积水;②双肾实质回声增强。入院后患儿持续无尿,解血性尿,肾功能急剧恶化,血小板持续下降。于是我们立即给予以下对症支持处理:置温箱保暖,止血,输液及静脉营养,保护脏器,维持血糖、电解质稳定等。同时输入同型红细胞悬液,多巴胺及呋塞米对症处理血尿,碳酸氢钠碱化尿液。入院第3天红细胞计数3.68×1012/L(下降),血红蛋白120 g/L(下降),血小板计数47×109/L(下降),网织红细胞(%)8.51%(上升)。生化:谷丙转氨酶63 U/L(升高),谷草转氨酶138 U/L(升高),总胆红素119.0 μmol/L(升高),直接胆红素4.9 μmol/L,间接胆红素114.1 μmol/L(升高),尿酸1018.9 μmol/L(升高),BUN 9.87 mmol/L(升高),肌酐280 μmol/L(升高),乳酸脱氢酶超过1867 U/L,碳酸氢根13.7 mmol/L(下降),钾6.35 mmol/L(升高),钠120.4 mmol/L(下降),氯86.5 mmol/L(下降),总蛋白42.6 g/L(下降),钙1.44 mmol/L(下降),磷2.58 mmol/L(升高)。第4天BUN 10.50 mmol/L(升高),肌酐402 μmol/L(上升)。
胸部正位片:双肺纹理增强。颅脑彩超检查诊断:①左侧丘脑、颞顶枕叶及右侧顶叶脑实质内异常回声(考虑大范围出血,伴部分区域小囊腔形成);②左侧室管膜下出血破入脑室;③右侧脑室增宽;④大脑动脉血流阻力减低,怀疑脑水肿。患儿出现急性肾功能衰竭后,立即在原治疗基础上予以持续性肾脏替代治疗及血浆置换:持续性肾脏替代治疗和血浆置换使用Fresenius multiFiltrate,B超引导下行右侧颈内静脉置管,使用BRAUN 5F双腔中心静脉导管,B超定位导管尖端位于右心房水平,采用multiFiltrate kit Paed血液透析滤过器及管路套包,滤器型号为Ultraflux AV Paed,膜面积0.2 m2,容量18 mL,管路容量为54 mL。第1天行连续性静脉-静脉血液透析(continuous venous venous hemodialysis,CVVHD)治疗,5%白蛋白预充管路及滤器,血流速度16 mL/min,透析液速度为300 mL/h,枸橼酸抗凝。第2天行首次血浆置换(plasma exchange PE)+CVVHD治疗,5%白蛋白预充管路及滤器,血浆置换采用血浆分离器为Plasmaflo OP-02W(膜面积为0.2 m2,容量25 mL),血流速度10 mL/min,分浆速度150 mL/h,肝素抗凝,血浆置换量150 mL,约为患儿血浆量的1.2倍,血浆置换治疗结束后更换血浆分离器为血液透析滤过器,继续行CVVHD治疗,前置换,置换液速度190 mL/h,下机后等量鱼精蛋白中和。肾脏替代治疗和血浆置换后第2天血常规:红细胞计数1.81×1012/L(下降),血红蛋白58.0 g/L(下降),血小板计数93.0×109/L(下降);生化:谷丙转氨酶21 U/L,谷草转氨酶36 U/L,总胆红素116.3 μmol/L(升高),直接胆红素6.0 μmol/L,间接胆红素110.3 μmol/L(升高),尿酸335.0 μmol/L,BUN 2.40 mmol/L,肌酐177 μmol/L(升高),乳酸脱氢酶1312 U/L,碳酸氢根27.3 mmol/L,肌酸激酶94 U/L,钾4.48 mmol/L,钠143.4 mmol/L,氯103.9 mmol/L,钙2.12 mmol/L,磷1.60 mmol/L。
结合患儿病史及辅助检查结果,诊断为:①新生儿非典型溶血性尿毒综合征;②新生儿急性肾功能衰竭;③新生儿重度贫血;④新生儿血小板减少症;⑤右肾积水;⑥电解质紊乱;⑦低蛋白血症;⑧肝功能异常;⑨新生儿肺炎;⑩新生儿黄疸;早产儿脑病;颅内出血;早产适于胎龄儿(孕35周+1,BW 2.48 kg);低出生体重儿(BW 2.48 kg)。
追问病史,患儿父亲、母亲健康,无特殊疾病史。非近亲婚配。母亲妊娠第35周+1时发现一胎胎心停止,立即行剖宫产。为明确诊断,经医学伦理审核以及家长知情同意,采集患儿血液分别送往院外医学检验研究所检测补体水平,乙二胺四乙酸(EDTA)抗凝管送至基因公司行基因分析。补体检测结果CFH 1451.33(参考值210.0~425.5),CFHR水平1257.83(参考值265.5~1292.5),CHI 441.44(参考值42.5~288.5),补体C3转化酶抗体816.5(参考值95~538)。这表明补体替代途径被异常激活。采用全外显子组检测基因拷贝数变异(copy number variation,CNV)分析,制备DNA文库,使用全外显子测序(whole exome sequencing,WES)高通量测序平台检测基因外显子区域及旁侧内含子区域,将测序结果与人类基因组hg19参考序列进行对比,当检测出致病或者可能致病变异存在于常染色体隐性基因中时,通过高通量测序技术(next-generation sequencing technology,NGS)测序确保该基因编码序列的覆盖率达100%。检测发现与患儿临床症状相关的长达57430 bp大片段杂合变异,未检测出可以解释临床表型的单核苷酸变异或微小缺失/重复变异。该致病性杂合缺失位于1号染色体q31.3区(seq[GRh37]1q31.3),涉及CFHR1和CFHR3基因的大片段杂合变异。CFHR3、CFHR1和CFHR4是连续基因,以这种相对顺序出现在1号染色体上的1q31~q32.1处。这些基因是补体激活簇中的调节因子。每个包含6个外显子,与分泌蛋白的生成有关[6]。补体检测结果中CFH浓度显著升高,与基因结果符合,亦支持了aHUS诊断。CFHR1和CFHR3的纯合缺失,通常伴有CFHR滴度升高[7],而本例为杂合缺失,所以CFHR滴度不高。我们对患儿父母行全基因组CNV分析,未检测到与先证者有关的变异。因为该技术检测的是全基因组范围内大于100 kb的染色体缺失、微重复,而父亲的突变基因长度只有数个10 kb。于是对父母CFHR3/CFHR1基因采用多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)进行验证和家系分析,检测发现父亲CFHR3基因和CFHR1基因外显子区域存在大片段杂合缺失,未发现其母CFHR3/CFHR1外显子区域存在大片段缺失/重复,如图1。为求进一步验证又进行全基因组拷贝数变异检测(SNP array),发现母亲X染色体q13.1 q21.1区域发生约7.3 Mb拷贝数中性杂合缺失[arrXq13.1q21.1(71,607,449~78,942,721)×2],但目前尚未见与之有关疾病的相关文献报道,故考虑为非致病性突变。父亲未检测出染色体拷贝数异常。
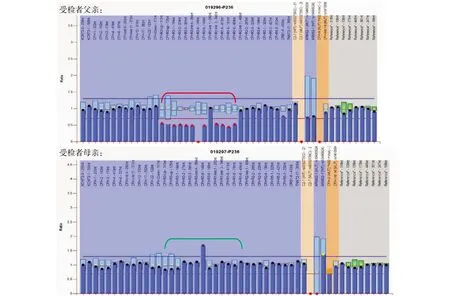
CFHR3-6-148nl和CFHR1-Intrl-282nl这2个探针易受其他多态性影响,建议忽略这2个探针的结果
2 讨论
患儿系同卵双胎之一,另一胎宫内死亡,入院时血红蛋白低,血小板低,全身苍白,皮肤可见散在出血点。入院后解鲜红色血尿,无尿,贫血,血小板减少且在输血纠正后继续下降,同时肾功能急剧恶化,我们怀疑HUS,请儿童肾脏专科协助诊疗后也一致认为患儿应考虑诊断该病。因患儿感染指标不高,我们认为感染的可能性不大,病因可能是先天性基因及补体途径异常所致,立即对患儿检测血清补体水平及全外显子组CNV分析。基因检测结果显示患儿及其父亲在1号染色体q31.3区均检测出杂合缺失,患儿出生即发病,父亲为携带者,未发病。双胎中另一胎比该患儿发病更早,所以直接导致宫内死亡。在本病例处理中,将危及患儿生命的肾功能衰竭放在治疗的首要地位,积极治疗肾衰,纠正严重贫血,维持呼吸循环的稳定,保护并减少器官的进一步损伤,获得较好的疗效。在明确临床诊断后予以肾脏替代治疗2次及血浆置换1次。治疗后复查患儿肾功能逐渐恢复,恢复自行排尿,但患儿头部B超显示严重颅内出血,家属放弃治疗。
HUS是一种以溶血性贫血伴碎片细胞、血小板计数低和急性肾功能衰竭3种特征表现为主的的血栓性微血管疾病。HUS往往起病急,病情发展快,易反复。大多发病于学龄期及学龄前期儿童,在新生儿期发病较为罕见[8-9]。
aHUS是非腹泻性及共患病性HUS,具有预后差、致死率高的特点,发病年龄范围很大,大多在婴儿期发病,发病第二高峰出现在25岁至40岁,通常由妊娠诱发补体激活失控导致全身血管内皮损伤,继发血栓形成[8-10]。各系统广泛受累,肾脏损害最常见,约60%的患者会发展成终末期肾病,肾移植后1年肾脏存活率下降近半数[11]。aHUS患者中60%检测到先天性补体替代途径失调,这表明了该病的遗传性和家族聚集性。aHUS在儿童(<20岁)中的发病率约为(0.25~0.75)/107[8,10,12]。
2.1 病理机制
在aHUS中,补体系统异常导致补体沉积于内皮细胞,使内皮细胞肿胀及脱落,小动脉和毛细血管增厚,继发血液中的成分聚集并吸附在细胞表面导致切割红细胞的机械性血栓形成,产生分裂细胞,继而形成了非免疫性溶血性贫血、肾功能障碍、血小板减低三联征[13-14]。肾内膜细胞是补体激活和免疫复合物沉积的主要场所,所以肾脏是最常见的受累器官[15]。
补体系统是通过经典途径,凝集素途径和替代途径帮助宿主清除病原体。替代途径使C3在触发因素下激活成为C3b,与CFB和CFD共同作用形成C3转化酶,CFH、CFI、MCP可将其灭活。当CFH、CFB、CFI、MCP基因突变,CFB过度活化,CFH、CFI、MCP灭活C3b的作用下降,C3b激活大量C3转化酶,使得C5大量激活成为C5b,最终大量激活的膜攻击复合体(membrane attack complex,MAC)导致微血管血栓病[15],如图2所示。
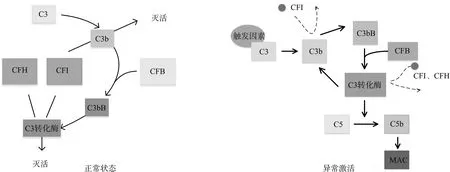
图2 补体调节系统正常及异常激活状态
CFH及其5种相关蛋白(CFHR1~CFHR5)的基因位于1号染色体上长度为355 kb的着丝粒片段中。对该区域的序列分析证明CFH和CFHR1~CFHR5之间具有高度同源性,易发生转化和非等位同源重组[3]。CFHR1/CFHR3缺失导致血浆对蛋白保护活性降低,导致补体激活调节异常。变异后的调节因子活性大幅下降,但也可能发生积极性变异(保护性变异)对抗消极性变异。补体激活取决于调节蛋白积极性变异和消极性变异的总和,轻微的缺陷不容易表现出来。相反,较重的补体调节系统缺陷就很容易被触发(如感染、癌症、药物、妊娠、自身免疫或器官移植)。约有30% aHUS发病机制还不明确。补体调节失调在其他类型的溶血性尿毒综合征中也有报道。
2.2 诊断
TMA主要包括aHUS、TTP、志贺毒素相关性HUS和其他伴有全身性疾病性TMA。这些疾病的相似性使鉴别诊断的难度大大增加。与TMA相关的全身性疾病包括妊娠期HELLP综合征(溶血伴肝酶升高和血小板计数低)、全身性感染(艾滋病病毒、巨细胞病毒等)、肿瘤、药物、系统性红斑狼疮、移植等。对于感染性腹泻相关性HUS的诊断,粪便常规及培养可鉴别产志贺毒素大肠埃希菌(STEC),血培养菌落结果可鉴定肺炎链球菌,鼻咽拭子PCR可鉴别甲型流感。钴胺素C缺乏相关性HUS可通过检测出高血清同型半胱氨酸和低蛋氨酸诊断[10]。ADAMTS13活性低于10%,极有可能是TTP,临床表现以伴有神经系统症状为特点[10]。DGKE基因变异者大多在生后数年发病。
严格来说TMA的诊断应该是通过组织活检作出的病理诊断,然而在临床中可通过观察到血小板减少症、非免疫介导溶血性贫血来推断出TMA。一旦确定了TMA的诊断,就必须区分志贺毒素相关性HUS、TTP、aHUS和其他全身疾病性TMA。
感染性腹泻相关性HUS的诊断是基于临床或实验室诊断腹泻病,或者曾是感染性腹泻暴发的暴露者,大便中大肠埃希菌或痢疾志贺菌微生物培养阳性可确诊,通过免疫测定检测志贺毒素也很重要。检测为阴性或者缺乏腹泻病史及临床表现可考虑其他疾病的诊断。ADAMTS13活性分析是鉴别TTP和aHUS的关键,活性<10%高度提示TTP。ADAMTS13活性分析通常需要一定时间,这就需要一些快捷的诊断方法,研究表明,严重ADAMTS13缺乏的患者通常表现为血肌酐<200 μmol/L,血小板计数<30×109/L[11];约50%的患者还伴有抗核抗体阳性。存在共患疾病情况下极有可能是继发性HUS。
aHUS是在排除以上疾病后考虑的。具体诊断指标为:血红蛋白<100 g/L,血小板<150×109/L;外周血涂片有破碎红细胞碎片,网织红细胞升高,Coombs’试验阴性,乳酸脱氢酶升高(超过460 U/L,通常处于非常高的水平,反映了血管溶血及弥漫性组织缺血);同时存在急性肾损伤,即血肌酐水平较同年龄同性别水平有1.5倍升高[14]。
aHUS患者需要对补体途径进行相对全面的评估。包括C3、CFH、CFI、CFB、MCP和CFHR的血清滴度。本病例还检测了C3转化酶滴度。接着用二代基因检测C3,CD46(MCP),CFB,CFH,CFHR1,CFHR3,CFHR4,CFI,DGKE和CFHR的基因。简易诊断流程如图3。
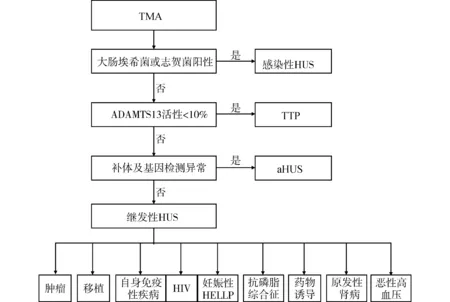
图3 aHUS的鉴别诊断
2.3 治疗
支持性治疗:严重贫血时输注血细胞,一般不需要输血小板,除非血小板计数<10×109/L、有活动性出血或者进行侵入性手术。液体和电解质的管理是维持血管内容积状态和避免多系统器官衰竭的关键。应及时纠正电解质紊乱,避免使用肾毒性药物。适度使用药物处理高血压。出现尿毒症、液体超负荷和电解质紊乱情况下应进行肾脏替代治疗。血浆置换和补体抑制剂Eculizumab提供专业治疗。
血浆置换和血浆输注(plasma exchange and plasma infusions,PE/PI):尽管儿童可能出现相关技术并发症,血浆置换和血浆输注是几乎唯一在全球各地均可获得的治疗方法,仍是aHUS的重要治疗方法。它可以去除患者体内的抗体、多种蛋白及毒素等,但不能解决补体功能不全的根本原因。方法:通常前5 d至少每天交换血浆体积的1.2倍,60 mL/kg,直到连续2 d血小板>100×109/L,之后隔天进行1次,然后每周2次。血小板计数和血清乳酸脱氢酶(LDH)浓度是监测血浆疗法最敏感指标。血浆治疗应持续到血小板计数和血清LDH浓度保持正常为止[10]。
Eculizumab是一种单克隆人源化抗C5抗体,可阻止C5裂解和C5a和C5b-9的形成,从而阻断MAC的形成[16]。特别适用于PE无效或PE依赖的预后较差的aHUS患儿。这种治疗对无论有无补体突变的患者都有效。但孤立性DGKE突变的患者不建议使用。其副作用是阻断了补体激活,使患者感染的易感性增加,因此强烈建议用药前2周接种脑膜炎奈瑟菌疫苗[13,16],并提前保护性应用抗生素。尽管有耐药的报道,甲基青霉素仍被推荐用于预防脑膜炎球菌。建议儿童接种流感嗜血杆菌疫苗和肺炎链球菌疫苗。2009年,Eculizumab首次应用于aHUS病例,现已在美国和欧盟地区被批准用于aHUS的治疗,中国大陆地区尚待批准引进。
活体器官移植:CFH、CFI、CFB和C3突变的患者因为复发风险高,不建议器官移植。禁忌亲属作为供者。肾移植对腹泻型HUS效果确切[17]。MCP突变患者移植通常有良好的预后。CFHR阳性者,尽早通过血浆交换清除CFHR,可以提高移植后器官存活率[14]。CFH和CFI是由肝脏产生的,因此,对于肾功能保留的患者,若治疗条件不足,可提前主动选择肝移植甚至肝肾联合移植[18]。
新治疗方法:针对补体级联反应中过度活化分子的抗体如C5,C3的治疗方法正在临床试验中,血浆纯化或者重组CFH和CFI用来替代自身功能不足的因子[18]。
综上所述,非典型溶血性尿毒综合征是一种罕见的、预后差、致命性的疾病,是由补体替代途径的过度激活导致调节功能异常引起的。对于新生儿难以纠正的贫血、血小板减少、急性肾功能衰竭要考虑到该病。在积极对症治疗的同时,有必要积极完善基因检测,以明确诊断,减少误诊及漏诊,便于及早干预,评估预后及指导遗传咨询,降低疾病发病率和病死率。
猜你喜欢
家教世界(2022年34期)2023-01-08 13:52:50
昆明医科大学学报(2021年8期)2021-08-13 09:00:02
昆明医科大学学报(2021年2期)2021-03-29 07:42:24
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
中国医药指南(2017年3期)2017-11-13 02:55:32
兽医导刊(2016年6期)2016-05-17 03:50:31
实用肝脏病杂志(2015年5期)2015-12-03 06:28:07
百科知识(2015年18期)2015-09-10 07:22:44
当代畜禽养殖业(2014年12期)2014-02-27 08:00:10
