
Duplication of 19q (13.2-13.31) associated with comitant esotropia: A case report
2021-08-04 05:25YueLanFengNingDongLi
World Journal of Clinical Cases 2021年20期
Yue-Lan Feng, Ning-Dong Li
Yue-Lan Feng, Ning-Dong Li, Department of Ophthalmology, National Center for Children's Health, Beijing Children’s Hospital, Capital Medical University, Beijing 100045, China
Yue-Lan Feng, Department of Ophthalmology, First Hospital Affiliated to Baotou Medical College, Baotou 014010, Inner Mongolia Autonomous Region, China
Abstract BACKGROUND Comitant esotropia is the most common form of strabismus. It is caused by heterogeneous environmental and genetic risk factors. The pure duplication of the long arm of chromosome 19 is a rare abnormality. Only 8 patients with partial trisomy of the long arm of chromosome 19q have been reported to date. Here, we describe a girl with pure duplication of 19q, who was diagnosed with congenital esotropia, microcephaly, and gallbladder agenesis.CASE SUMMARY The patient was diagnosed with esotropia when she was 1-year-old. The Krimsky method showed +50 prism diopters in the primary gaze position. No additional abnormal findings were observed following slit lamp and fundus examination,but the features of the full-field electroretinogram showed a decreased amplitude and increased implicit times. Magnetic resonance imaging showed ventriculomegaly with thinning of the corpus callosum and splenium in her brain. A 4.42 Mb mosaic duplication within 19q13.2-q13.31 region (chr19:39,343,725 to 43,762,586) was detected by microarray comparative genomic hybridization.CONCLUSION Strabismus is reported in many live borns with pure duplication of 19q. This important clinical characteristic indicates that the candidate genes fundamental for this phenotype may be narrowed to genes within the 19q13.3-q13.31 region.There were two candidate genes observed that may contribute to the comitant esotropia phenotype, namely XRCC1 (19:43,543,311) and SMG9 (19:43,727,991).
Key Words: Duplication; 19q; Esotropia; Strabismus; XRCC1; SMG9; Case report
INTRODUCTION
Comitant esotropia is a very common form of childhood strabismus with a prevalence of approximately 2.5% among White populations of European ancestry and 0.5%among Africans and Asians[1]. It is frequently noted in infancy or early childhood as the angle of esotropia misalignment between the two eyes remains relatively constant with changes in gaze direction. Comitant esotropia is often accompanied by amblyopia(uniocular visual neglect), a leading cause of visual impairment in children and young adults.
The pathogenesis of comitant esotropia remains largely unknown. Although many studies based on family trio and twins have demonstrated a genetic contribution to strabismus[2-5], there are limited data supporting Mendelian segregation. Parikhet al[6] reported the results of linkage analysis in a large family with non-syndromic strabismus with presumed autosomal recessive inheritance, which has been identified as the first susceptibility locus on chromosome 7p22.1[6]. Another study on 55 Japanese families including at least two members with comitant strabismus revealed that the loci at chromosomes 4q28.3 and 7q31.2 showed a significant evidence of linkage[7], which focuses on the chromosomal loci down toWNT2andMGST2[8].Beyond this, the genetic contributions to comitant strabismus remain undefined.
Duplication of 19q is a rare chromosome abnormality that may affect a number of genes. To date, only 8 patients with partial trisomy of 19q have been reported[9-16](Table 1). We found that strabismus was present in some of these cases, indicating that the pathogenic genes fundamental for the strabismus phenotype might be associated with chromosome 19q duplications.
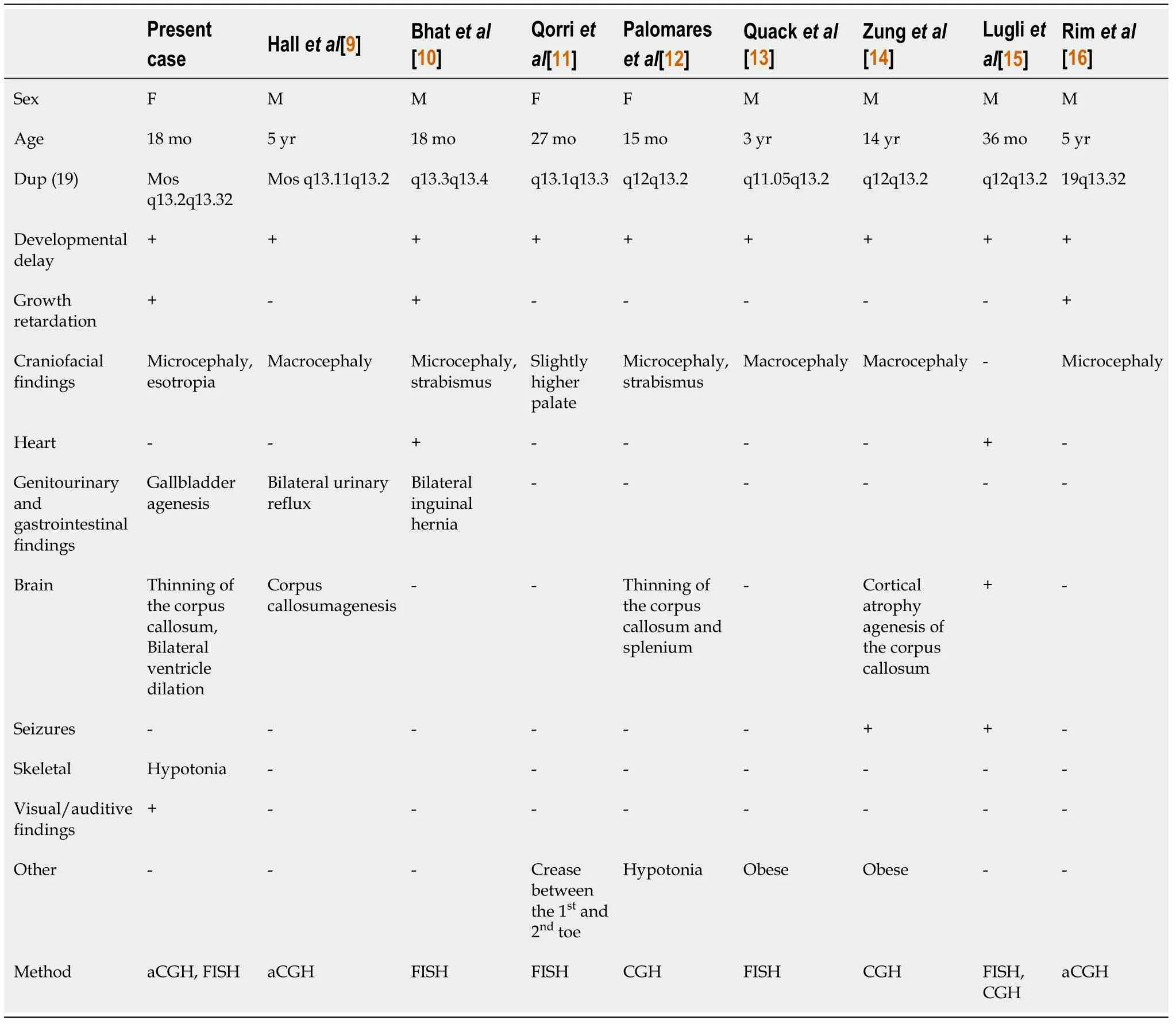
Table 1 Summary of clinical features comparing live borns with trisomy 19q
We present a girl with 19q duplication (13.2-13.31), who was diagnosed with microcephaly, comitant esotropia, developmental delay, and gallbladder agenesis. We used conventional G-band karyotyping and array comparative genomic hybridization(aCGH) to explore the potential correlation between the phenotype and the genetic disorder.
CASE PRESENTATION
Chief complaints
An 18-mo girl accompanied by her father and mother, presented to the Ophthalmology Department in August 2018. Her parents complained about her strabismus for more than 6 mo (Figure 1A).

Figure 1 The examination of the patient. A: Patient has microcephaly and congenital esotropia; B: Corpus callosum and splenium are thin; C: Ventricle is dilated.
History of present illness
The parents complained about her strabismus.
History of past illness
The patient was born at 39 wk as the first fetus of her healthy, non-consanguineous parents and was deliveredviaCesarean section. When she was born, the Apgar score was 6 at 1 min and 8 at 5 min. She was diagnosed with microcephaly and develop-mental delay because her body weight, length, and head circumference were 2140 g(3rdpercentile), 43 cm (3rdpercentile), and 30 cm (25thpercentile), respectively. She was also found to have gallbladder agenesis upon B-scanning ultrasonic examination.
Personal and family history
The absence of this duplication in her parents involved in the chromosomal and aCGH analyses indicated that the 4.42 Mb duplication was ade novorearrangement in this affected patient.
Physical examination
The Krimsky method showed +50 prism diopters in the primary gaze position.Cycloplegic refraction showed +3.50 D sph in the right eye and +3.25 D sph in the left eye. No additional abnormal findings were observed following slit lamp and fundus examination, but the features of the full-field electroretinogram (FF-ERG) showed a decreased amplitude and increased implicit times.
Laboratory examinations
A normal karyotype (46, XX) was revealed at the 550-band resolution using the conventional G-band karyotyping with her peripheral blood (Figure 2). However, a 4.42-Mb mosaic duplication within the 19q13.2-q13.31 region (chr19:39,343,725 to 43,762,586 ) was detected by aCGH (MACArray Karyo 1440 BAC-chip; Macrogen,Seoul, Korea) (Figure 3), which was further confirmed by fluorescencein situhybridization using the RP11-264N23 (19q13.2) probes (Empire Genomics LLC, Buffalo, NY,United States) on both interphase and metaphase spreads presenting a mosaic ratio of 92% (46 cells) in 50 examined cells and only 8% (4 cells) with a normal karyotype(Figure 4). The absence of this duplication in her parents involved in the chromosomal and aCGH analyses indicated that the 4.42 Mb duplication was ade novorearrangement in this affected patient.
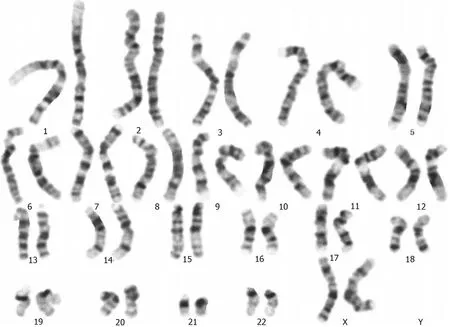
Figure 2 G-banded analysis at the 550-band level shows a normal karyotype, 46, XX (20).

Figure 3 Array comparative genomic hybridization analysis performed suggests a mosaic gain of 19q. A: Array comparative genomic hybridization (aCGH) data profile in whole chromosomes. A dot represents a bacterial artificial chromosome clone, the X-axis represents the chromosome number (1-22, X,Y), and the Y-axis represents the log2 T/R signal ratio value. The table below the graph represents the average log2 T/R signal ratio value for each chromosome. Green dots represent a copy number gain (log2 T/R signal ratio value > 0.25) and duplication on chromosome 19; B: aCGH profile from chromosome 19 shows a duplication on the long arm. The size of the duplication fragment was estimated to be 4.42 Mb (chr19:39,343,725-43,762,586).
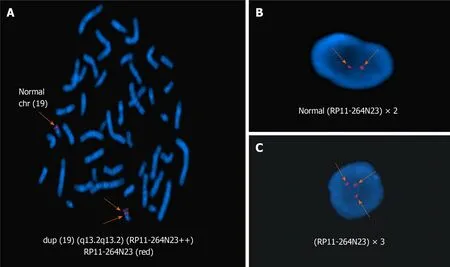
Figure 4 Fluorescence in situ hybridization with the 19q13.2-specific region probe (red). A: Arrow indicates a dup (19) (q13.2q13.2) chromosome[46,XX.ish dup (19) (q13.2q13.2) (RAI1++)] in metaphase fluorescence in situ hybridization (FISH); B: In interphase FISH, two arrows indicate a normal signal probe(RP11-264N23) × 2; C: In interphase FISH, three arrows indicate a duplication of the probe (RP11-264N23) × 3.
Imaging examinations
Magnetic resonance imaging showed ventriculomegaly with thinning of the corpus callosum and splenium in her brain (Figure 1B and C).
FINAL DIAGNOSIS
The final diagnosis of the presented case was congenital esotropia, microcephaly, and gallbladder agenesis.
TREATMENT
She underwent bilateral medial rectus recession of 6.0 mm at the age of 18 mo. Forced duction tests were performed intraoperatively and showed no mechanical force or restriction of the extraocular muscles.
OUTCOME AND FOLLOW-UP
Her postoperative measurement was 10Δ esotropia (Krimsky) after 1 mo. Extraocular muscles movement were normal.
DISCUSSION
Pure duplications of 19q are rare, and as previously reported in the literature, only 8 cases were live born. To our knowledge, the patient described in this study is the second confirmed case of mosaicism involving duplication of the 19q region. In 2010,Hallet al[9] reported the first case of mosaic trisomy in 19q13.11q13.2 with obesity,macrocephaly, and global developmental delay. Psychomotor or mental retardation is a common feature in these live borns. In addition, microcephaly was described in 3 of 8 cases[9,13,16]. Three patients had alterations in the corpus callosum[9,12,14],consistent with our case. The duplication region 19q13.2-13.31 in this case contained 27 OMIM Morbid genes (Figure 5), but only Plekhg2 is currently known to have a phenotype associated with leukodystrophy and acquired microcephaly. Edvardsonet al[17] reported 5 children from two unrelated consanguineous Palestinian families with a severe neurodevelopmental disorder. The children presented in infancy with delayed psychomotor development, hypotonia, and postnatal progressive microcephaly. Brain imaging of all patients showed a similar pattern of abnormal white matter, consistent with leukodystrophy. Finally, it was inferred that abnormality of pleckstrin homology domain containing, family G member 2 expression might cause microcephaly, abnormal white matter, and developmental delay. Thus, it is not surprising that our patient presented with those clinical features, and our findings evidently further confirm the genotypic and phenotypic links of this gene abnormality[18,19].

Figure 5 Genomic coordinates for the 19q13.2-q13.31 gain are chr19:39,343,725-43,762,586 (hg38), estimated to be 4.42 Mb. This duplication includes 27 OMIM Morbid genes: Plekhg2, Timm50, Dll3, Dyrk1b, Akt2, Pld3, Prx, Sptbn4, Ltbp4, Coq8b, Itpkc, Cyp2a4, Cyp2a5, Cyp2b10, Tgfb1, B9d2, Bckdha,Rps19, Cd79a, Atp1a3, Erf, Cic, Megf8, Lipe, Ethe1, Xrcc1, and Smg9 (hg38 database, http://genome.ucsc.edu).
Comitant esotropia is another special feature that was described in 3 of these live born. Bhatet al[10] reported the first case of smallde novoduplication of 19q (13.3-13.4)with comitant esotropia, and our patient was also found to have esotropia in 19q (13.2-13.31). These important clinical characteristics may indicate that the candidate genes fundamental for these phenotypes could be narrowed to the genes within the 19q13.3-q13.31 region. There were two candidate genes observed that may contribute to the comitant esotropia phenotype,XRCC1(19:43,543,311) andSMG9(19:43,727,991).XRCC1 is a molecular scaffold protein that assembles multiprotein complexes involved in DNA single-strand break repair[10]. The XRCC1 protein complexes are important for normal neurological function. Hochet al[20] showed that biallelic mutations in human XRCC1 were associated with ocular motor apraxia, axonal neuropathy, and progressive cerebellar ataxia. TheSMG9gene encodes an essential component of nonsense-mediated mRNA decay. Shaheenet al[21] reported two unrelated consanguineous families of Arab origin in which 5 patients had a heart and brain malformation syndrome, including hypertelorism, small eyes, and poor vision.Further analyses of the gene network showed that two genes (ubiquitin A-52andribosomal protein S27A) can either interact with XRCC1 or SMG9, which are important candidates associated with comitant esotropia, suggesting the potential roles of their interactions during eye development (Figure 6).
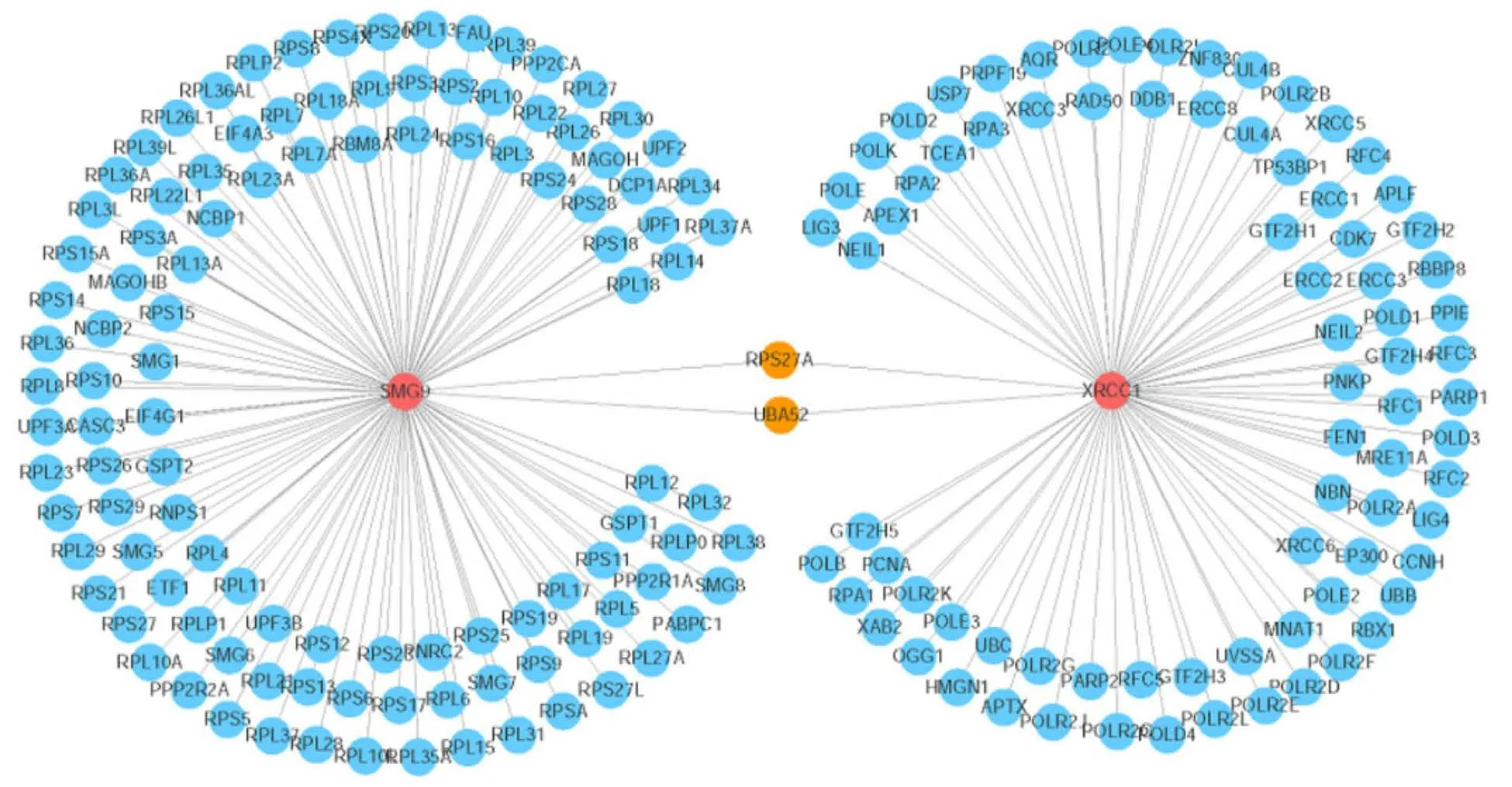
Figure 6 Visualization of the gene network that interacts with the SMG9 and XRCC1 genes. The red circles indicate the query genes (SMG9 and XRCC1), the orange circles represent the genes that interact with both of the two query genes, and the blue circles denote the other interacting genes. The line thickness indicates the strength of the interaction (interaction score).
Although it is still unclear how the copy number change affects the actions ofXRCC1andSMG9or their interactions, the genetic evidence shows that these two genes are associated with eye development, and the possible duplication may determine an overexpression of some or all of these genes, leading to an imbalanced eye development.
CONCLUSION
To conclude, the present patient provides further support for a distinct 19q duplication phenotype comprising developmental delay, microcephaly, thinning of the corpus callosum, and esotropia. The other phenotypic features are more variable, such as gallbladder agenesis and the moderately subnormal results of the FF-ERG stimulus threshold testing, which were not observed in previous case reports. There is a possibility that mosaicism may, in itself, cause the phenotype rather than the sole effect of 19q anomalies. Clinical evaluation of additional patients will be required to further delineate this phenotype.
 World Journal of Clinical Cases2021年20期
World Journal of Clinical Cases2021年20期
- World Journal of Clinical Cases的其它文章
- Obesity in people with diabetes in COVID-19 times: Important considerations and precautions to be taken
- Revisiting delayed appendectomy in patients with acute appendicitis
- Detection of short stature homeobox 2 and RAS-associated domain family 1 subtype A DNA methylation in interventional pulmonology
- Borderline resectable pancreatic cancer and vascular resections in the era of neoadjuvant therapy
- Esophageal manifestation in patients with scleroderma
- Exploration of transmission chain and prevention of the recurrence of coronavirus disease 2019 in Heilongjiang Province due to inhospital transmission