
Potential treatment with Chinese and Western medicine targeting NSP14 of SARS-CoV-2
2021-07-20 01:28ChoLiuXioxioZhuYiyoLuXinqinZhngXuJiTiYng
Cho Liu,Xioxio Zhu,Yiyo Lu,Xinqin Zhng,b,Xu Ji,*,Ti Yng
aNon-coding RNA and Drug Discovery Key Laboratory of Sichuan Province,Chengdu Medical College,Chengdu,Sichuan,610500,China
bBasic Medical School,Chengdu Medical College,Chengdu,Sichuan,610500,China
cSchool of Pharmacy,Chengdu Medical College,Chengdu,Sichuan,610500,China
Keywords:
SARS-CoV-2
Nonstructural protein 14(NSP14)
ZINC database
Drug docking
A B S T R A C T
The outbreak of coronavirus disease 2019(COVID-19)caused by severe acute respiratory syndrome coronavirus 2(SARS-CoV-2)is a serious global health threat.This raises an urgent need for the development of effective drugs against the deadly disease.SARS-CoV-2 non-structural protein 14(NSP14)carrying RNA cap guanine N7-methyltransferase and 3′-5′exoribonuclease activities could be a potential drug target for intervention.NSP14 of SARS-CoV-2 shares 98.7% of similarity with the one(PDB 5NFY)of acute respiratory syndrome(SARS)by ClustalW.Then,the SARS-CoV-2 NSP14 structures were modelled by Modeller 9.18 using SARS NSP14(PDB 5NFY)as template for virtual screening.Based on the docking score from AutoDock Vina1.1.2,18 small molecule drugs were selected for further evaluation.Based on the 5 ns MD simulation trajectory,binding free energy(ΔG)was calculated by MM/GBSA method.The calculated binding free energies of Saquinavir,Hypericin,Baicalein and Bromocriptine for the N-terminus of the homology model were-37.2711± 3.2160,-30.1746± 3.1914,-23.8953 ± 4.4800,and -34.1350 ± 4.3683 kcal/mol,respectively,while the calculated binding free energies were-60.2757±4.7708,-30.9955±2.9975,-46.3099±3.5689,and-59.8104±3.5389 kcal/mol,respectively,when binding to the C-terminus.Thus,the compounds including Saquinavir,Hypericin,Baicalein and Bromocriptine could bind to the N-terminus and C-terminus of the homology model of the SARS-CoV-2 NSP14,providing a candidate drug against SARS-CoV-2 for further study.
1.Introduction
The emergence of severe acute respiratory syndrome coronavirus 2(SARS-CoV-2)has caused a large global outbreak and is a major public health issue.The high Affinity of Spike protein for binding to human ACE2 protein makes SARS-CoV-2 spread rapidly in the population[1,2].Importantly,the death rate caused by coronavirus disease 2019(COVID-19)is much higher than the SARS outbreak in 2002-2003[3].The World Health Organization(WHO)declared that COVID-19,a pandemic disease caused by SARS-COV-2,has spread to more than 200 countries around the world.However,there is currently no effective drug or vaccine approved to treat COVID-19.
The SARS-CoV-2 genome is a positive-sense,single-stranded mRNA,which contains a 5′-cap structure and a 3′-poly(A)tail[4].The large replicase polyproteins 1a(pp1a)and pp1b,which are encoded by the 5′-terminal open reading frame 1a/b(ORF1a/b)at the 5′-terminus of the CoV genome,are cleaved by viral proteases to produce non-structural proteins(NSPs)such as RNA-dependent RNA polymerase(RdRp)and helicase(NSP13)[5].In addition,the non-structural proteins 14(NSP14)plays an important role in the virus’genome replication and transcription,and it is generally considered as an important functional protein in the coronavirus family[6,7].NSP14 is a bifunctional enzyme carrying S-adenosyl methionine(SAM)dependent(guanine-N7)N7-methyl transferase(N7-MTase)and 3′-5′exoribonuclease(ExoN)activities[5,8].The C-terminal domain N7-MTase activities involved in RNA cap modification,the cap structure at the 5′end of viral mRNA assists in translation and evading host defense[9].The N-terminal 3′-5′ExoN plays a proofreading role in the prevention of lethal mutagenesis[5,8],which provides an attractive target for drug design.Computational protein-ligand docking and virtual drug screening could accelerate the discovery of the potential drugs as potential candidates against COVID-19.Compounds based on virtual screening,such as ribavirin,lopinavir and ritonavir,have been shown to inhibit SARS-CoV-2 replication in vitro[10-13].In this work,the protein homology modeling and molecular docking were performed to select drug candidates targeting NSP14 to treat COVID-19.
2.Materials and methods
2.1.Homology modeling
The SARS-CoV-2 amino acid sequence(Accession number:MN908947)was obtained from database of the National Center for Biotechnology Information(NCBI).The SARS NSP14 amino acid sequence was downloaded from the PDB protein structure database(PDB ID:5NFY).The homology of above amino acid sequence was aligned using ClustalW and rendered using ESPript version 3.0.Homology model of the target protein was constructed and optimized by Modeller 9.18 using crystal structure of SARS NSP14(PDB ID:5NFY)as template.A total of 100 independent structures were constructed,and the one with best DOPE score was selected for further energy minimization by Amber.
2.2.Docking method
The previous study reported that NSP14 crystal structure of SARS is binocular,amino acids 1-287 fold into the ExoN domain,amino acids 301-527 form the N7-MTase domain[8].In addition,amino acids 288-301 form a complex loop structure connecting ExoN domain and N7-MTase domain.The ExoN domain shares a similar two-metal-ion-assisted mechanism for removal of misincorporated nucleotides[14,15],and the position of Mg2+is also considered to be the active center of exonuclease[8].Therefore,the docking box was set in a pocket formed by amino acid residues(D90,V91,E191,G189,H188,A187,W186,T277 and D273)around Mg2+(Fig.1).The docking box of N7-MTase of NSP14 was set where the substrates GpppA and SAM are located(Fig.1)[8],and the key amino acids were organized in Table S1.
The ligands were downloaded from the ZINC database(FDA,world-not-FDA, investigational-only, http://zinc.docking.org/substances/subsets/).The 2D structure of the compound was then converted into the corresponding 3D coordinates using the Babel server(http://openbabel.sf.net).Then the model was converted to pdbqt format by prepare_receptor4.py script with assigning atom type and partial charge.All rotatable bonds in the ligands were set as flexible for flexible docking.Vina1.1.2 was used for molecular docking.
2.3.Binding free energy calculation
Each simulation system was immersed in a cubic box of TIP3P water with 10 Å distance from the solute.The Na+or Cl-was applied to neutralize the system.General Amber force field(GAFF)15 and Amber ff14SB force field were used to parameterize the ligand and protein respectively.Altogether 10,000 steps of minimization with constraints(10 kcal/mol/Å2)on heavy atoms of complex,consisting of 5000 steps of steepest descent minimization and 5000 steps of conjugate gradient minimization,were used to optimize each system.Then each system was heated to 300 K within 0.2 ns,followed by 0.1 ns equilibration in NPT ensemble.Finally,5 ns MD simulation on each system at 300 K was performed.The minimization,heating and equilibrium were performed with sander program in Amber18.The 5 ns production run was performed with pmemd.cuda.Based on the 5 ns MD simulation trajectory,binding free energy(ΔG)was calculated with MM/GBSA method according to the following equation:
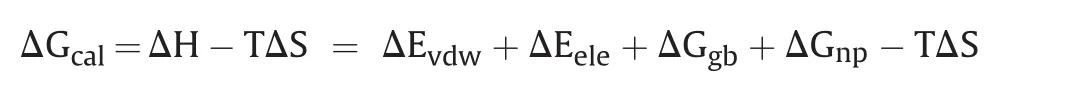
where ΔEeleand ΔEvdwrefer to electrostatic and van der Waals energy terms,respectively,ΔGgband ΔGnprefer to polar and nonpolar solvation free energies,respectively.Conformational entropy(TΔS)was not calculated for saving time.Besides,the ligands were compared based on the same target,so it is reasonable to ignore the entropy.
3.Results and discussion
3.1.Sequence and structural analyses
The SARS-nCoV-2 of NSP14 amino acid sequence downloaded from Protein BLAST was very conservative(data not shown).The amino acid sequence alignment revealed that the NSP14 of SARSCoV-2 shared 98.7% of similarity with the NSP14(PDB ID:5NFY)of SARS(Fig.S1),indicating that the results of NSP14 homology modeling would be more reliable.From the 100 homologous structures constructed,the one with the highest DOPE score was selected for the next drug docking.The two active sites of NSP14 were respectively set with two docking pockets(Fig.1).
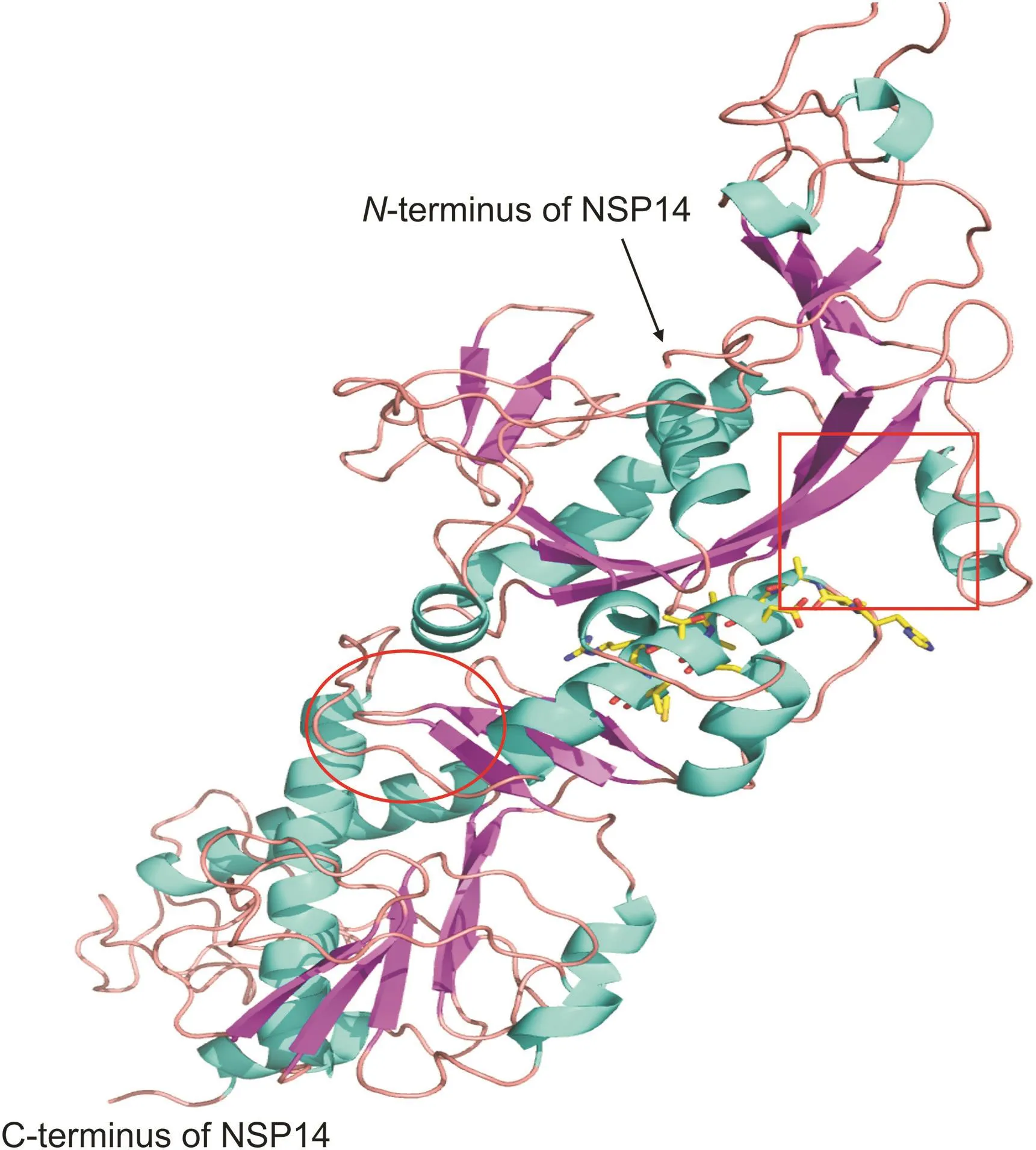
Fig.1.Cartoon representation of NSP14 homology modeling structure;the active pockets for drug docking are set in red ellipses(C-term)and squares(N-term).
3.2.Drug docking and screening
Ten top compounds that showed the lowest negative vina score in a range of-8.6 to-9.7 kcal/mol were selected from the N-terminal domain of homology model(Table 1),and eight top compounds with lowest negative vina score in a range of-8.7 to-9.7 kcal/mol were achieved from the C-terminal domain of homology model(Table 2).It was unexpectedly discovered that the four compounds might simultaneously combined the N-terminal and C-terminal active centers of the NSP14 simulated structure.These four compounds were Saquinavir,Hypericin,Baicalein and Bromocriptine,respectively.
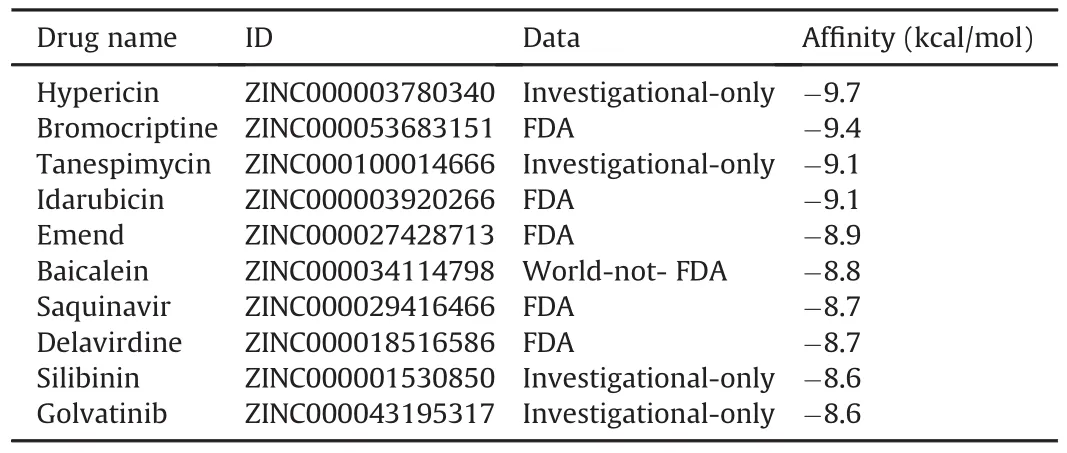
Table 1 Ten drugs selected from the N-terminal domain of homology model.
3.3.Docking results of Saquinavir against SARS-CoV-2 NSP14 model
Saquinavir,as the first FDA-approved human immunodeficiency virus(HIV)protease inhibitor,has been used in the treatment of HIV patients since 1995[16].Saquinavir is safe and generally welltolerated in HIV-1-infected adults[17].Five of the hydrogen bonds involving ASP-273,ASN-252,ASP-90,ALA-187 and LEU-253 were maintained upon the binding of Saquinavir and N-terminus of SARS-CoV-2 NSP14 based on our docking results(Fig.2A).Meanwhile,hydrogen bonds involving ASN-386,GLN-313,GLY-333 and THR-428 maintained upon the binding of Saquinavir and C-terminus of SARS-CoV-2 NSP14(Fig.2C).Saquinavir could bind to the N-and C-terminal active pockets of the SARS-CoV-2 NSP14(Figs.2B and D).The recent study from a drug-target interaction deep learning model showed that Saquinavir can bind to SARS-CoV-2 RNA-dependent RNA polymerase to inhibit the enzyme activity[18].Our simulation results showed that Saquinavir could bind to two active sites of NSP14;thus Saquinavir could be a candidate drug against SARS-CoV-2 for further research.
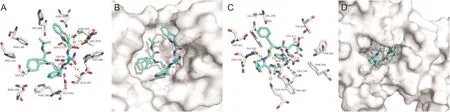
Fig.2.The binding model of Saquinavir against SARS-CoV-2 NSP14.(A)Interactions between Saquinavir(cyan)and associated residues(off-white)in the N-terminus of the homology model for SARS-CoV-2;(B)Binding models of Saquinavir(cyan)in the SARS-CoV-2 NSP14 protein N-terminus pocket(white surface);(C)Interactions between Saquinavir(cyan)and associated residues(off-white)in the C-terminus of the homology model for SARS-CoV-2;(D)Binding models of Saquinavir(cyan)in the SARS-CoV-2 NSP14 protein C-terminus pocket(white surface).Numbers accompanying dashed yellow lines represent the interaction distance(Å).
3.4.Docking results of Hypericin against SARS-CoV-2 NSP14 model
Hypericin,a main ingredient of traditional Chinese medicine Hypericum perforatum L.(St.John's wort),has demonstrated the activity against RNA viruses in vitro by inhibiting viral replication[19].The present docking results showed that three of the hydrogen bonds involving ASN-252,GLY-93,and HIS-268 were maintained upon the binding of Hypericin and N-terminus of SARS-CoV-2 NSP14(Fig.3A).The eight hydrogen bonds involving ASN-306(double),ARG-310(double),ASN-422,TYR-420 and LYS-336(double)were maintained upon the binding of Hypericin and C-terminus(Fig.3C).Hypericin could bind to the N-and C-terminal active pockets of the SARS-CoV-2 NSP14 as well(Figs.2B and D).Hypericin has been proven to have inhibitory effects on human hepatitis C virus(HCV)and human immunodeficiency virus(HIV)[20].Combined with the present study,Hypericin may have a potential antiviral effect against SARS-CoV-2.Shuanghuanglian oral liquid,composed of Hypericum perforatum L,has been widely used for the treatment of viral influenza.However,Shuanghuanglian oral solution suggested for the treatment of SARS-CoV-2 has triggered a huge crisis of public trust among Chinese scientists.We suggested that anti-SARS-CoV-2 effects of Hypericin should be examined in cell culture models with SARS-CoV-2 infection.This will help us have a good understanding of whether it is a good or not to use Chinese medicines for the treatment of SARS-CoV-2.
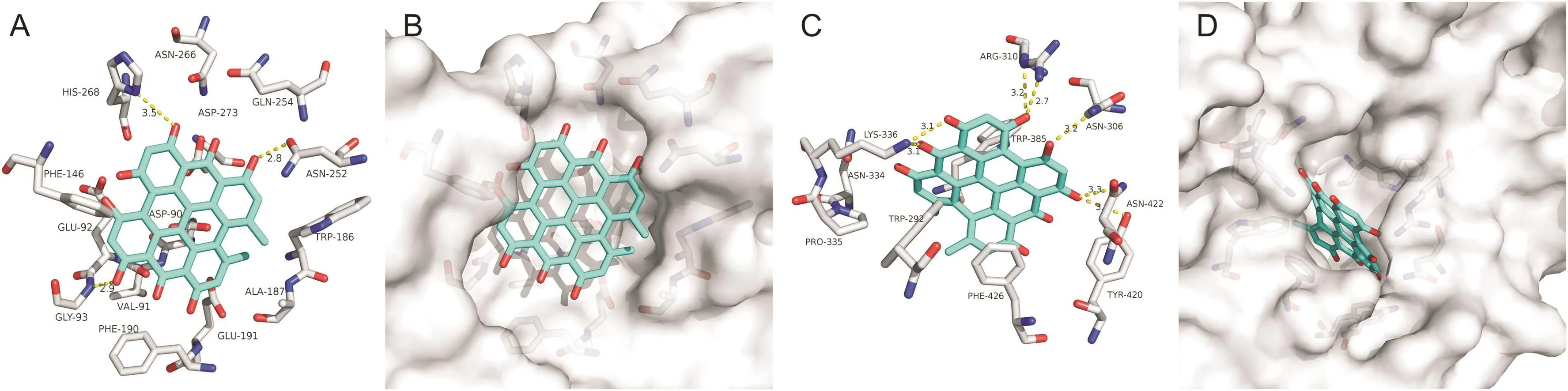
Fig.3.The binding model of Hypericin against SARS-CoV-2 NSP14.(A)Interactions between Hypericin(cyan)and associated residues(off-white)in the N-terminus of the homology model for SARS-CoV-2;(B)Binding models of Hypericin(cyan)in the SARS-CoV-2 NSP14 protein N-terminus pocket(white surface);(C)Interactions between Hypericin(cyan)and associated residues(off-white)in the C-terminus of the homology model for SARS-CoV-2;(D)Binding models of Hypericin(cyan)in the SARS-CoV-2 NSP14 protein C-terminus pocket(white surface).Numbers accompanying dashed yellow lines represent the interaction distance(Å).
3.5.Docking results of Baicalein against SARS-CoV-2 NSP14 model
Baicalein,a flavonoid compound isolated from the root of Scutellaria baicalensis Georgi(Huang Qin in Chinese),inhibits viral replication of parainfluenza,influenza A,hepatitis B,HIV-1,and SARS coronavirus[21-23].The present docking results showed that seven of the hydrogen bonds involving ASN-266,ASN-252,ASP-273,GLY-93,GLU-92 and HIS-268(double)were maintained upon the binding of Baicalein and N-terminus of SARS-CoV-2 NSP14(Fig.4A).Five hydrogen bonds involving ASN-386(double),ASP-331(double),and GLN-313 were maintained upon the binding of Baicalein and C-terminus(Fig.4C).Baicalein could also bind to the N-and C-terminal active pockets of the SARS-CoV-2 NSP14(Figs.4B and D).The previous study showed that Baicalein as a novel chemical inhibitor could inhibit ATPase activity of NSP13 protein of SARS coronavirus[24].The present data suggested that Baicalein may bind to NSP14 protein to exert anti-SARS-CoV-2 activity.Therefore,we suspect that the anti-SARS-CoV-2 activity induced by Baicalein could be valuable for further study.
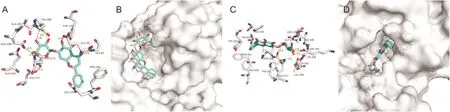
Fig.4.The binding model of Baicalein against SARS-CoV-2 NSP14.(A)Interactions between Baicalein(cyan)and associated residues(off-white)in the N-terminus of the homology model for SARS-CoV-2;(B)Binding models of Baicalein(cyan)in the SARS-CoV-2 NSP14 protein N-terminus pocket(white surface);(C)Interactions between Baicalein(cyan)and associated residues(off-white)in the C-terminus of the homology model for SARS-CoV-2;(D)Binding models of Baicalein(cyan)in the SARS-CoV-2 NSP14 protein C-terminus pocket(white surface).Numbers accompanying dashed yellow lines represent the interaction distance(Å).
3.6.Docking results of Bromocriptine against SARS-CoV-2 NSP14 model
Bromocriptine,a specific dopamine receptor agonist for the hypothalamus and pituitary,has an inhibitory effect on replication of the Dengue virus with low cytotoxicity(half maximal effective concentration,EC50= 0.8-1.6 μM;and half maximal cytotoxicity concentration,CC50= 53.6μM)[25].Moreover,Bromocriptine inhibited Zika virus protease activities and exhibited synergistic effects with interferon-α2b against Zika virus replications[26].It is interesting that Bromocriptine could bind to the N-and C-terminal active pockets of the SARS-CoV-2 NSP14(Figs.5B and D).Three of the hydrogen bonds involving ASN-104,ASP-273 and GLN-145 were maintained upon the binding of Bromocriptine and N-terminus of SARS-CoV-2 NSP14(Fig.5A).There was one bond involving THR-428 maintained upon the binding of Bromocriptine and C-terminus of it(Fig.5C).
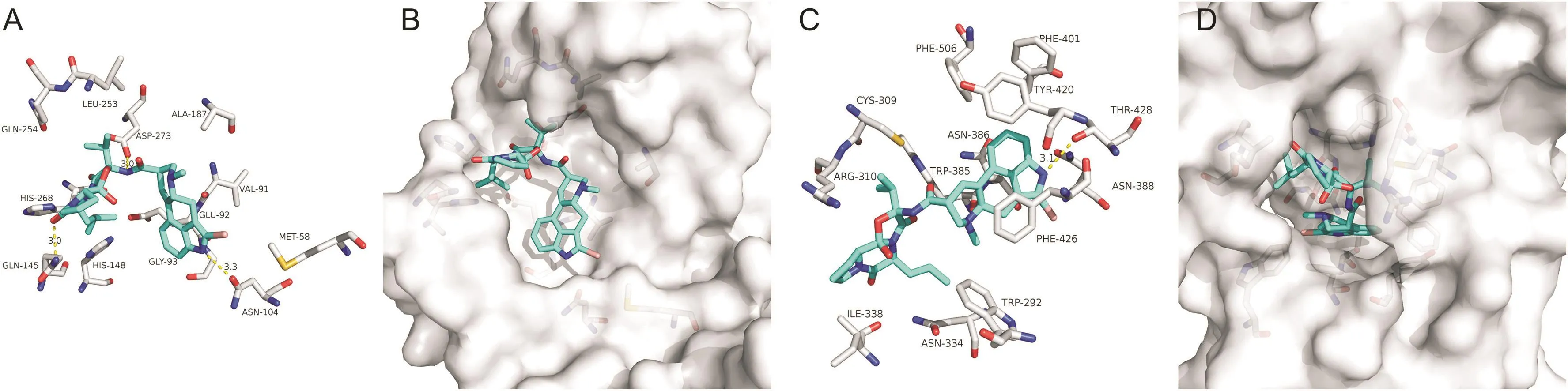
Fig.5.The binding model of Bromocriptine against SARS-CoV-2 NSP14.(A)Interactions between Bromocriptine(cyan)and associated residues(off-white)in the N-terminus of the homology model for SARS-CoV-2;(B)Binding models of Bromocriptine(cyan)in the SARS-CoV-2 NSP14 protein N-terminus pocket(white surface);(C)Interactions between Bromocriptine(cyan)and associated residues(off-white)in the C-terminus of the homology model for SARS-CoV-2;(D)Binding models of Bromocriptine(cyan)in the SARS-CoV-2 NSP14 protein C-terminus pocket(white surface).Numbers accompanying dashed yellow lines represent the interaction distance(Å).
3.7.Binding free energy
The Table S2 indicates that Saquinavir,Hypericin,Baicalein and Bromocriptine interact with key amino acid in the active center of NSP14.Based on the 5 ns MD simulation trajectory,ΔG was calculated by MM/GBSA method.The calculated binding free energies of Saquinavir,Hypericin,Baicalein and Bromocriptine for the N-terminus of the homology model were-37.2711±3.2160,-30.1746±3.1914,-23.8953±4.4800,and-34.1350±4.3683 kcal/mol,respectively(Table 3),while the calculated binding free energies were-60.2757± 4.7708,-30.9955± 2.9975,-46.3099±3.5689,and-59.8104±3.5389 kcal/mol,respectively,when binding to the C-terminus(Table 4).Taken together,the results demonstrated that Saquinavir had a strong binding free energy.
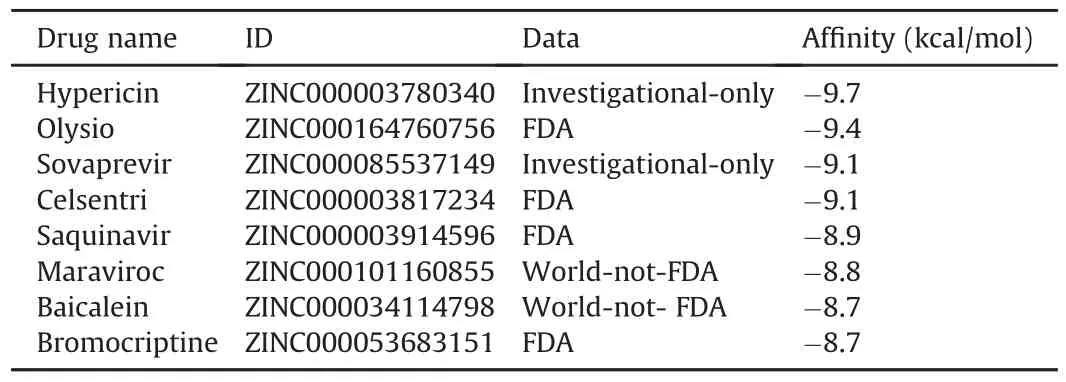
Table 2 Eight drugs selected from the C-terminal domain of homology model.
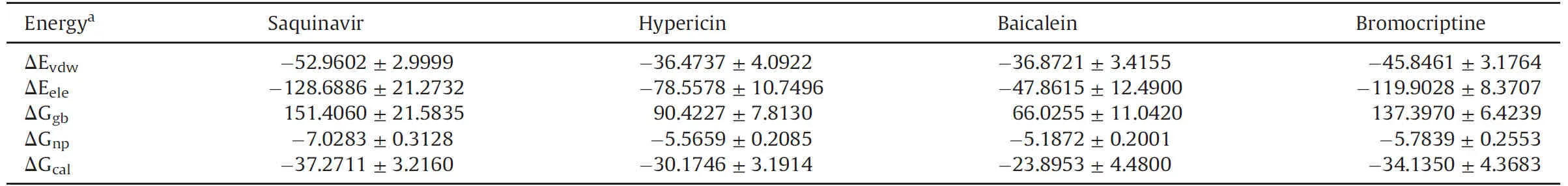
Table 3 The calculated binding energies of ligand to the N-terminus of SARS-CoV-2 NSP14.
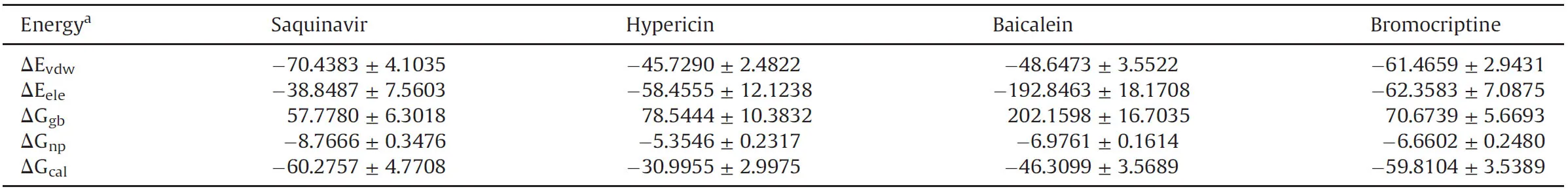
Table 4 The calculated binding energies of ligand to the C-terminus of SARS-CoV-2 NSP14.
4.Conclusion
SARS-CoV-2 NSP14,a bifunctional enzyme carrying RNA cap guanine N7-methyltransferase and 3′-5′exoribonuclease activities,could be a potential drug target for intervention.SARS-CoV-2 NSP 14 shares 98.7% of sequence similarity with the corresponding one in SARS.Thus,the homology model of SARS-CoV-2 NSP14 was structured for virtual screening.Based on the docking score,18 drugs were selected for further evaluation.Four drugs(Saquinavir,Hypericin,Baicalein and Bromocriptine)could bind to the N-terminal and C-terminal domains of SARS-CoV-2 NSP 14,and these drugs all interacted with key amino acid residues in the active center.We suggest the anti-SARS-CoV-2 effects of the above four drugs should be evaluated in the cell culture models with SARSCoV-2 infection.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China(Grant Nos.31870135,31600116)and the “1000 Talent Plan”of Sichuan Province(No.980).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2020.08.002.
 Journal of Pharmaceutical Analysis2021年3期
Journal of Pharmaceutical Analysis2021年3期
- Journal of Pharmaceutical Analysis的其它文章
- Molecular detection of SARS-CoV-2 being challenged by virus variation and asymptomatic infection
- The potential of miRNA-based therapeutics in severe acute respiratory syndrome coronavirus 2(SARS-CoV-2)infection:A review
- Accurate and sensitive determination of hydroxychloroquine sulfate used on COVID-19 patients in human urine,serum and saliva samples by GC-MS
- Fast saccharide mapping method for quality consistency evaluation of commercial xylooligosaccharides collected in China
- Dispersive liquid-liquid microextraction,an effective tool for the determination of synthetic cannabinoids in oral fluid by liquid chromatography-tandem mass spectrometry
- Identification and quantification of the bioactive components in Osmanthus fragrans roots by HPLC-MS/MS