
四嗪类化合物理化性质的理论研究
2021-07-19 09:36陈鑫鑫李春香张裕平崔乘幸屈凌波
河南科技学院学报(自然科学版) 2021年4期
陈鑫鑫李春香张裕平崔乘幸,屈凌波
(1.河南科技学院化学化工学院,河南新乡453003;2.郑州大学化学学院,河南郑州450052)
含能材料是一类在外界能量刺激下能够发生自氧化还原反应并释放大量能量的化合物或混合物,广泛应用于军事、航天和民用领域,如推进剂火药、猛炸药和起爆药等爆炸物[1-3].含能材料的核心是不同种类的具有高能量密度的含能化合物,如唑类、立方烷类、嗪类等[4-6].常用氧平衡来衡量高能化合物爆破效果和爆炸后生成有害气体量,最理想的是零氧平衡.1,2,3,4-四嗪-1,3-二氧类化合物具有高达49%的含氮量,其四嗪环上的N-氧基团的两个氧原子能够提高其稳定性[7-8].当四嗪环上的两个氢原子被-NO2或-NF2取代时,可以获得氧平衡接近于零的衍生物.因此,该类化合物近年来得到该领域研究者的广泛关注并合成了大量的新型衍生物[8].
含能化合物的合成和性能研究通常十分危险,对于处于设计和评估阶段的目标化合物,其性能往往无法通过试验手段进行研究,因此在设计新型含能化合物时,理论与计算手段已经逐渐成为一种主流的研究方法.通过理论计算,科学家可以对设计的一系列分子的构型、电子结构、光学性质等进行较系统的研究,以了解其基本性能,为试验提供参考[9-11].如果一些含能化合物通过理论计算手段被认为是稳定的,就会极大地鼓舞化学家对这些分子的性质进一步展开研究;相反,当理论计算结果表明一些分子不能成为高能候选物时,则既能减少盲目实验造成的浪费与危险,又能缩短试验周期提高效益[12-16].
含能材料的结构和性质,归根结底是其电子结构决定.因此本文以1,2,3,4-四嗪-1,3-二氧为基本骨架,通过引入含能取代基(-NO2-、-NF2-、-NON2-或-C(NO2)3-)和桥基(-NH-或-N=N-)设计一系列衍生物[17].运用密度泛函理论,对其电子结构进行系统研究,并根据能量和稳定性相结合的标准,筛选出潜在的候选物.通过研究含能化合物的分子结构、键长、键角、二面角,探讨其结构以及它们的各种物理、化学、爆炸爆轰性能[18],对含能材料的分子设计、合成或生产、使用和贮存,都有重要的参考或指导作用[19-21].本研究使用最新的明尼苏达系列密度泛函方法MN15,对30种1,2,3,4-四嗪-1,3-二氧系列衍生物类化合物进行了理论计算,并与先前的理论计算和试验结果进行了对比,以期对该类化合物的新型衍生物设计提供一定的参考[22-23].
1 计算方法
30种四嗪类化合物的结构式如图1所示.

图1 30种四嗪类化合物的结构式Fig.1 The formula of 30 tetrazines studied
本文使用理论计算手段对图所示的30种1,2,3,4-四嗪-1,3-二氧化合物的理化性质进行了系统的理论计算.首先在MN15/6-311G(d,p)水平下对这些化合物的结构进行全优化,相同的理论计算水平下进行频率分析计算,以确保优化得到的结构为极小值点.基于试验结果,我们对图1所示的30种高能化合物的几何构型、电子结构、电荷分布以及红外光谱等性质进行了分析,并讨论了母体上的取代基对相关理化性质的影响.
2 结果与讨论
2.1 几何构型分析
在MN15/6-311G(d,p)水平下优化所得的30种四嗪类化合物的三维构型以及主要的构型参数所涉及的原子编号如图2所示.高能化合物a1-a15的主要结构参数见表1.

表1 高能化合物a1-a15的主要结构参数Tab.1 Main structural parameters of high energy compound a1-a15

图2 在MN15/6-311G(d,p)水平下优化所得的30种四嗪类化合物的三维构型以及主要的构型参数所涉及的原子编号Fig.2 The three dimensional configurations of 30 tetrazines optimized at the MN15/6-311G(d,p)level and the atomic numbers involved in the main configuration parameters
由表1可知,单环四嗪化合物中N1-C1的键长为1.328×10-10m,C1-C2的键长为1.369×10-10m,C2-N2的键长为1.373×10-10m,N1-C1-C2的键角为124.6°,C2-N2-N3的键角为121.6°.而a2-a15分别用H、NO2、NF2、ONO2、C(NO2)3等取代R1和R2,这些取代基为吸电子基亲电取代,其强度顺序为C(NO2)3>ONO2>NF2>NO2>H.与a1相比,a2的N1-C1的键长短0.016×10-10m,C1-C2的键长短0.007×10-10m,C2-N2的键长为0.006×10-10m,N1-C1-C2的键角大2.4°,C2-N2-N3的键角大0.4°.同理分析得知,试验所得N1-C2的键长为1.300×10-10~1.329×10-10m,C1-C2的键长为1.362×10-10~1.398×10-10m,C2-N2的键长为1.372×10-10~1.391×10-10m,N1-C1-C2的键角为123.4°~127.0°,C2-N2-N3的键角为119.5°~122.5°.其中被较强吸电子基取代的R1、R2的N1-C2的键长整体呈减小趋势,C1-C2的键长、C2-N2的键长、N1-C1-C2的键角整体呈上升趋势,C2-N2-N3的键角围绕原单环四嗪类化合物C2-N2-N3的键角上下波动.C2-N2键长最长,预示热解引发反应可能始于C2-N2键均裂,亦即C2-N2键可能是该类化合物的热解和起爆引发键.此外,C2-N2键有变长的趋势,相应的化合物则愈不稳定,相应的感度可能也就愈大.
高能化合物b1-b15的主要结构参数见表2.

表2 高能化合物b1-b15的主要结构参数Tab.2 Main structural parameters of high energy compounds b1-b15
由表2可知,杂环四嗪类化合物b1中N4-C5的键长为1.379×10-10m,C5-C6的键长为1.374×10-10m,C5-C6的键长为1.374×10-10m,C1-C2的键长为1.5×10-10m,C1-C2-N3的键角为115.34°,N4-C5-C6的键角为115.34°.R1分别用-、-NH-、-N=N-取代,其中-NH-为斥电子基亲核取代,R分别用H、NO2、NF2、ONO2、C(NO2)3取代.对于b1-b5,N4-C5、C5-C6、C2-N3和N4-C5的键长都比b1长,而C1-C2键长都比b1短.根据键长初步判断C1-C2键最长,可能是该类化合物的热引发键,其键长变长意味着稳定性降低.对于b6-b10,键长基本呈增大趋势,键角为减少趋势,而含N键的键长变长,预示着相应化合物越不稳定,相应的感度可能也就越大.而b11-b15中键长随着b11上下波动,键角和二面角呈减小趋势.以上很好的说明NO2、NF2、ONO2等对R1、R2的取代对四嗪类化合物稳定性的影响.
Chuang等人指出筛选高能密度化合物的引发键解离能需要高于80 kJ/mol[7].引入-NO2、-NF2、-ONO2、-C(NO2)3基有利于提高衍生物的爆轰性能,但却会降低其稳定性.基于爆轰性能、热稳定性和氧平衡,研究者提出在30种化合物中,图1所示的a2~a7、图2所示的b2、b3、b7、b8、b12、b13可被考虑作为HEDCs的候选物[8].a2~a7中与引发相关的主要结构参数为N1-C1和C2-N2的键长,N1-C1-C1和C2-N2-N3的键角.其中键长值从a2~a7分别是1.312×10-10m、1.312×10-10m、1.305×10-10m、1.320×10-10m、1.322×10-10m、1.310×10-10m和1.379×10-10m、1.380×10-10m、1.378×10-10m、1.374×10-10m、1.374×10-10m、1.382×10-10m,键角分别是127.0°、124.8°、125.8°、125.9°、123.7°、125.0°和122.0°、119.5°、120.7°、122.3°、120.7°、121.4°,由此可见,分子中键长呈减少趋势,意味着键能呈增大趋势,稳定性升高.从整体来看,四嗪环含氮骨架稳定性有所上升.而b系列与引发键相关的结构参数为N4-C5和C2-N3的键长;C1-C2-N3和N4-C5-C6的键角.其中N4-C5的键长分别为1.379×10-10m、1.391×10-10m、1.382×10-10m、1.378×10-10m、1.382×10-10m、1.382×10-10m、1.392×10-10m、1.373×10-10m、1.380×10-10m、1.388×10-10m、1.39×10-10m、1.392×10-10m、1.391×10-10m、1.386×10-10m、1.387×10-10m;C2-N3的键长分别为1.379×10-10m、1.392×10-10m、1.382×10-10m、1.426×10-10m、1.383×10-10m、1.382×10-10m、1.392×10-10m、1.373×10-10m、1.366×10-10m、1.423×10-10m、1.391×10-10m、1.399×10-10m、1.401×10-10m、1.396×10-10m、1.381×10-10m.由此可见,含氮键有变长的趋势,相应的分子则越不稳定,相应的感度可能有所提高.对比b系列分子的键长,其键长波动范围较大,可知不同取代基对该类分子的影响较大.
2.2 电子结构分析
前线轨道理论认为,有电子排布的能量最高的分子轨道(即最高占据轨道HOMO)和没有被电子占据的,和无电子排布的能量最低的分子轨道(即最低未占轨道LUMO)是决定一个体系发生化学反应的关键。HOMO能级的负值代表该物质的第一电离能,HOMO能级越高,电离能越低,说明分子越容易失去电子。LUMO为最低空轨道,其能级值越低,说明分子越容易得到电子。HOMO-LUMO能隙值(Egap)的大小反映了电子从占据轨道向空轨道跃迁的能力,故一定程度上代表分子参与化学反应及其对光反应敏感程度性的强弱.同时,HOMO-LUMO能隙值越大说明电子跃迁越不容易发生,则此分子就越稳定.
如表3所示为a1-a15的电子结构信息,所有的15种单环四嗪类化合物的电子态均为1-A.当单环未取代时,HOMO值为-8.60 eV,LUMO值为-2.78eV,|Gap|=5.82 eV;当引入一个含能取代基-NO2时,a2的HOMO值减小0.72 eV,LUMO值减小1.83 eV,|Gap|=4.71 eV,较a1减小了1.11 eV;当引入一个-NF2时,a5的HOMO值减小0.33eV,LUMO值减小0.31eV,|Gap|=5.84eV,较a1增加了0.014 eV;当引入-ONO2时,a8的HOMO值减小0.61 eV,LUMO值减小0.61 eV,|Gap|=5.82 eV;当引入一个-C(NO2)3时,a11的HOMO值减小0.66 eV,LUMO值减小1.96 eV,|Gap|=4.52 eV,较a1减小了1.30 eV.从表中数据可以发现,当1,2,3,4-四嗪-1,3-二氧分子中引入了含能取代基-NO2、-NF2、-ONO2、-C(NO2)3时,其HOMO和LUMO的能量水平与原分子相比会有所降低,稳定性也有所降低,而且-NO2降低的最多.C1上的Mulliken电荷在a10中最大,而C2上的电荷在a14中最大,这与其上的更多的吸电子基团有关.

表3 a1-a15对应的HOMO、LUMO、Egap、电子态、以及主要碳原子上的密立根电荷密度Tab.3 HOMO,LUMO,ΔE gap,electronic states,and the Milligan charge densities on major carbon atoms corresponding to a1-a15
b1-b15对应的HOMO、LUMO、Egap、电子态、以及主要碳原子上的密立根电荷见表4.a1-a15、b1-15的HOMO能量、LUMO能量以及HOMO-LUMO的带隙Egap的信息如图3所示.

图3 HOMO能量、LUMO能量以及HOMO-LUMO的带隙E gapFig.3 HOMO energy,LUMO energy,and HOMO-LUMO band gap EGAPNote
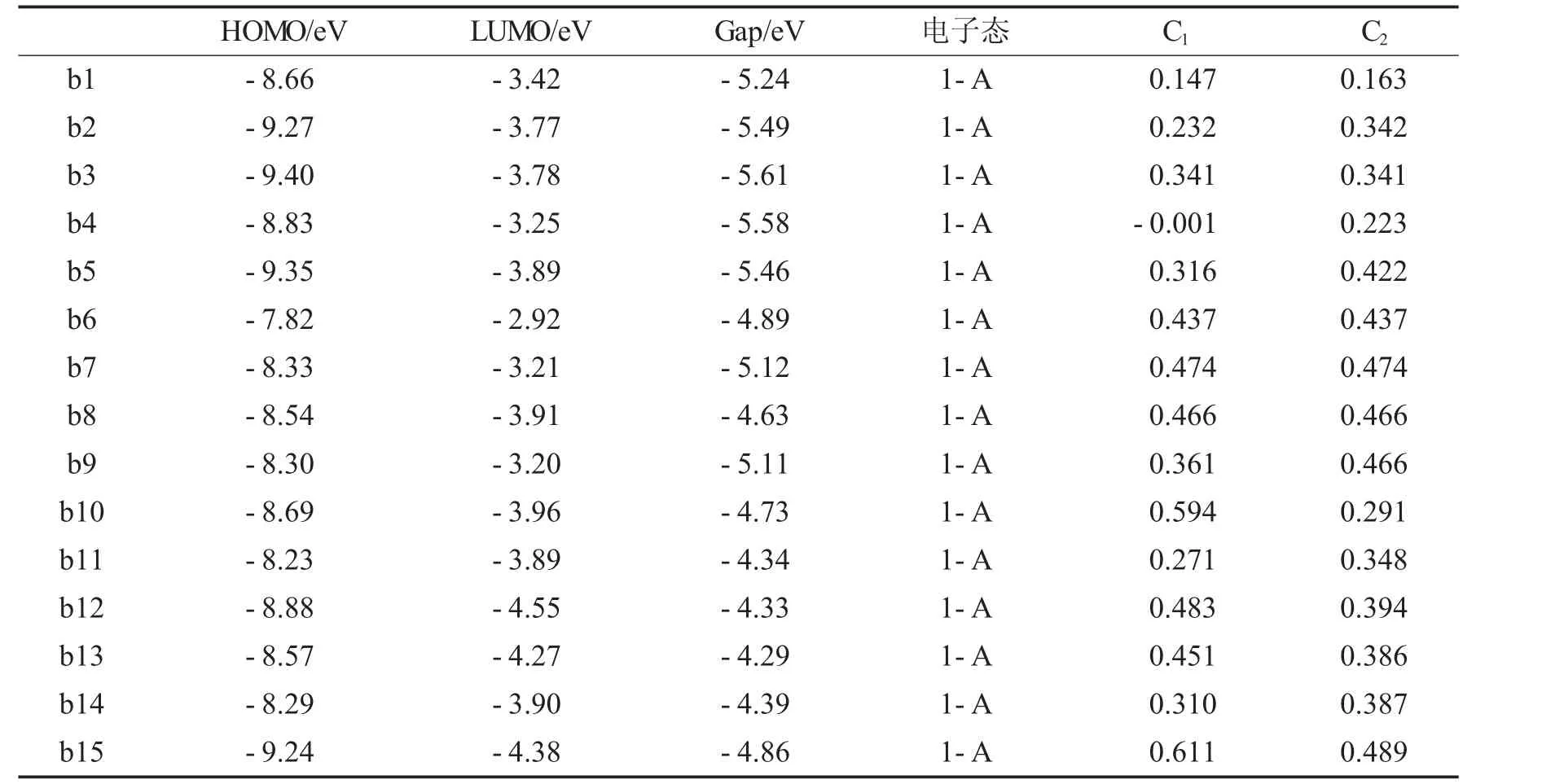
表4 b1-b15对应的HOMO、LUMO、Egap、电子态、以及主要碳原子上的密立根电荷Tab.4 HOMO,LUMO,Egap,electronic states,and Milligan charge densities on major carbon atoms corresponding to b1-b15
如表4可知,对桥基-NH-或-N=N-连接的双环化合物,引入与单环相同的取代基,15个衍生物的变化规律和单环类似,HOMO和LUMO值相对减小,能隙值相对减小,分子稳定性降低.从图三a)中(HOMO,LUMO与能隙折线图),我们观察能隙折线,可以发现在a5,a6,a7,a8,a9,a10处的能隙值比其他分子的能隙值更加突出,说明当取代基为-ONO2、-NF2时,电子从占据轨道向空轨道跃迁要困难些,也就是此衍生物的稳定性相对高于其它衍生物的稳定性.从图3-B观察到b1,b2,b3,b4,b5处的能隙值比其他分子的能隙值更加突出,说明当两环不以桥基相连时,电子从占据轨道向空轨道跃迁要困难些,此时衍生物的稳定性相对高于其它衍生物的稳定性.C1和C2上的Mulliken电荷在b7中最大,数值均为0.474.
2.3 红外光谱和振动分析
理论计算所得的30种高能化合物红外吸收强度最大振动模式对应的吸收波数见表5.

表5 理论计算所得的30种高能化合物红外吸收强度最大振动模式对应的吸收波数Tab.5 Absorption wave number corresponding to the maximum vibration mode of infrared absorption intensity of 30 high-energy compounds obtained by theoretical calculation
红外光谱是化合物的基本属性,也是分析和鉴别化合物的有效手段,化合物的红外光谱可用于计算其热力学性质.到目前为止,1,2,3,4-四嗪-1,3-二氧类衍生物已经被实验合成,但对该衍生物缺少理论研究,因此通过理论方法对其加以计算具有重要意义.表五给出的MN15/6-311G(d,p)水平下对设计的30种四嗪类衍生物的分子构型的几何构型全优化和频率,并在此初上得到的IR谱图中振动频率最大的吸收波数.由表中可见,1,2,3,4-四嗪-1,3-二氧类衍生物的红外吸收强度最大振动模式的吸收波数,主要集中在152 2.6~176 8.6 cm-1的波段处.作为1,2,3,4-四嗪-1,3-二氧类衍生物的特征区,此波段主要属于四嗪环骨架上的C=C反对称伸缩振动和四嗪环上N=O基团的对称伸缩振动.例如,表五化合物b14的红外吸收强度最大振动模式的吸收波数为164 5.59cm-1,对应四嗪环上N=O键,与化合物b1相比,N=O键反对称伸缩振动的频率移向高频区,出现蓝移现象.此外红外光谱上的157 2.3cm-1,159 1.6cm-1,166 8.2cm-1分别对应四嗪环上的其他三个N=O键.
3 结论
含能化合物由于其不稳定性,往往不能直接通过实验直接合成,并且含能化合物的热力学等性质的理论研究也十分有限,因此本文采用理论计算的方法,从热力学和动力学两方面对1,2,3,4-四嗪-1,3-二氧类衍生物的稳定性进行了研讨并对所研究的系列异构体的构型和电子结构等进行了分析与总结.从而探索了高能且具有较好热力学稳定性的高能化合物候选物,为试验合成与探究1,2,3,4-四嗪-1,3-二氧系列衍生物丰富了相关理论基础.由于本文所研究的1,2,3,4-四嗪-1,3-二氧类衍生物数量有限,因此对于该系列衍生物计算结果的分析、总结以及某些热力学性质的研究,还具有一定的局限性.在今后的研究工作中,我们可以通过试验的一些表征方法与理论计算结果进行有效的结合,并对通过分子设计所得到的更多的衍生物进行理论计算,然后对结果进行归纳总结,以期望对1,2,3,4-四嗪-1,3-二氧类的新型衍生物的研究提供更多的理论参考.
猜你喜欢
分子催化(2022年1期)2022-11-02
中草药(2022年9期)2022-05-06
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
湖北农业科学(2017年24期)2018-01-27
中学化学(2017年6期)2017-10-16
中学化学(2017年6期)2017-10-16
中学化学(2017年2期)2017-04-01
试题与研究·高考理综化学(2016年3期)2017-03-28
中学生数理化·高二版(2016年3期)2016-12-26
中学生数理化·高二版(2016年3期)2016-12-26
