
遗传性耳聋基因筛查在新生儿听力筛查中的应用价值
2021-07-13 02:19:50梁文英李永涛于晓静
新乡医学院学报 2021年6期
梁文英,李永涛,于晓静,王 丽
(1.洛阳市妇幼保健院新生儿科,河南 洛阳 471000;2.洛阳市妇幼保健院儿科,河南 洛阳 471000)
新生儿听力障碍是临床常见的先天性缺陷,主要指感音、传音等听力系统综合分析功能和神经中枢功能障碍及器质性病变导致的新生儿听力损伤或减退[1]。新生儿听力障碍不仅可造成耳聋,还严重影响患儿语言系统并导致新生儿性格向恶性方向发展,故需要及早诊断和干预。新生儿听力筛查(universal newborn hearing screening,UNHS)是临床早期发现新生儿听力障碍的主要方法,但针对初筛未通过的新生儿需要延迟至3个月后再进行复筛,并经过听力学诊断才可确诊,诊断周期需要3~6个月[2]。另外,针对大前庭水管综合征和药物敏感的新生儿,常规的听力筛查无法有效检出,需要结合其他方法明确患儿是否存在听力障碍。有研究显示,基因筛查对判断新生儿听力障碍具有较高的提示作用,亦可用于大前庭水管综合征和药物敏感的新生儿,是常规听力检查的良好补充[3]。因此,本研究旨在探讨遗传性耳聋基因筛查在新生儿听力筛查中的应用价值。
1 资料与方法
1.1 一般资料选择2018年10月至2019年10月在洛阳市妇幼保健院新生儿科出生的新生儿为研究对象。新生儿纳入标准:(1)均进行听力筛查和遗传性耳聋基因筛查;(2)父母民族均为汉族;(3)单胎妊娠;(4)新生儿监护人知情同意。排除标准:(1)有明显先天性耳部畸形;(2)中途失访。本研究共纳入新生儿3 887例,男2 011例,女1 876例;胎龄34~40(38.20±2.18)周,出生体质量2 320~4 680(3 150.55±510.03)g;剖宫产1 522例,自然分娩2 365例。本研究通过医院医学伦理委员会审核批准。
1.2 听力筛查新生儿出生3~5 d时进行听力初筛,听力初筛于噪音低于45 dB的相对安静环境进行。使用耳声发射仪(丹麦AccuScreen公司)进行耳声发射检查,使用全自动听性脑干反应测试仪(德国Maico公司)进行自动听性脑干反应(auto auditory brainstem response,AABR)检查;初筛患儿进行随访,于出生28~42 d进行复查;出生3个月内进行脑干听觉诱发电位测试[4]。
1.3 基因筛查新生儿出生72 h后取足跟血,将血液滴在基因筛查专用滤纸片,滤纸片晾干后提取基因组;将提取的DNA溶于生理盐水,按照耳聋基因筛查试剂盒(上海亿康医学检验所)说明书进行操作,检测GJB2、SLC26A4及12SrRNA等3个基因的突变位点,使用耳聋基因分析系统进行基因判断[5]。
1.4 统计学处理应用SPSS 22.0软件进行数据统计与分析,计数资料以例数和百分率表示,组间比较采用χ2检验,P<0.05为差异有统计学意义。
2 结果
2.1 耳聋基因筛查结果结果见表1。3 887例新生儿中,筛查出耳聋基因突变者201例,耳聋基因突变率为5.17%(201/3 887);其中235delC位点杂合突变占40.30%,IVS7-2A>G位点杂合突变占25.87%,2168A>G位点杂合突变占12.44%,299-300delAT位点杂合突变占11.44%,其余位点突变率均低于10.00%。
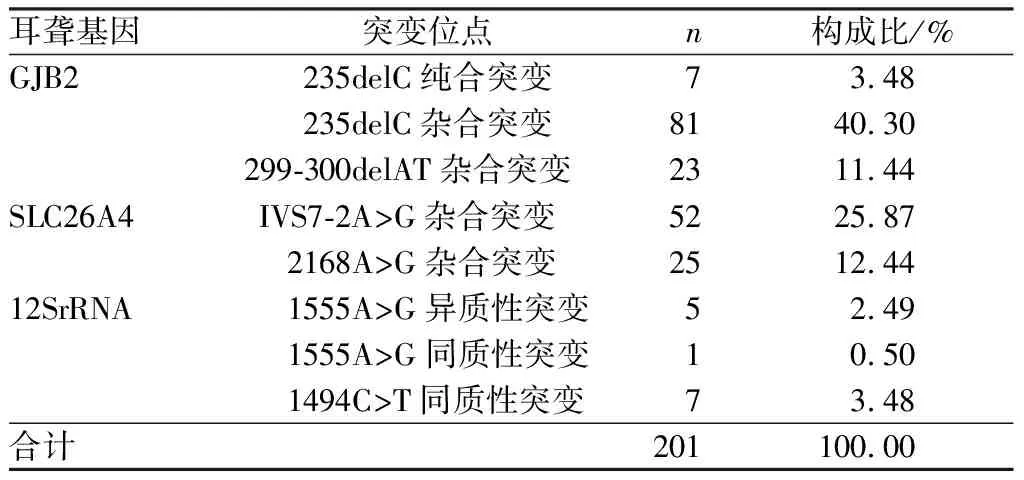
表1 201例新生儿耳聋基因突变情况
2.2 不同性别、胎龄、出生体质量及分娩方式新生儿耳聋基因突变率比较结果见表2。不同性别、胎龄、出生体质量及分娩方式新生儿耳聋基因突变率比较差异均无统计学意义(P>0.05)。
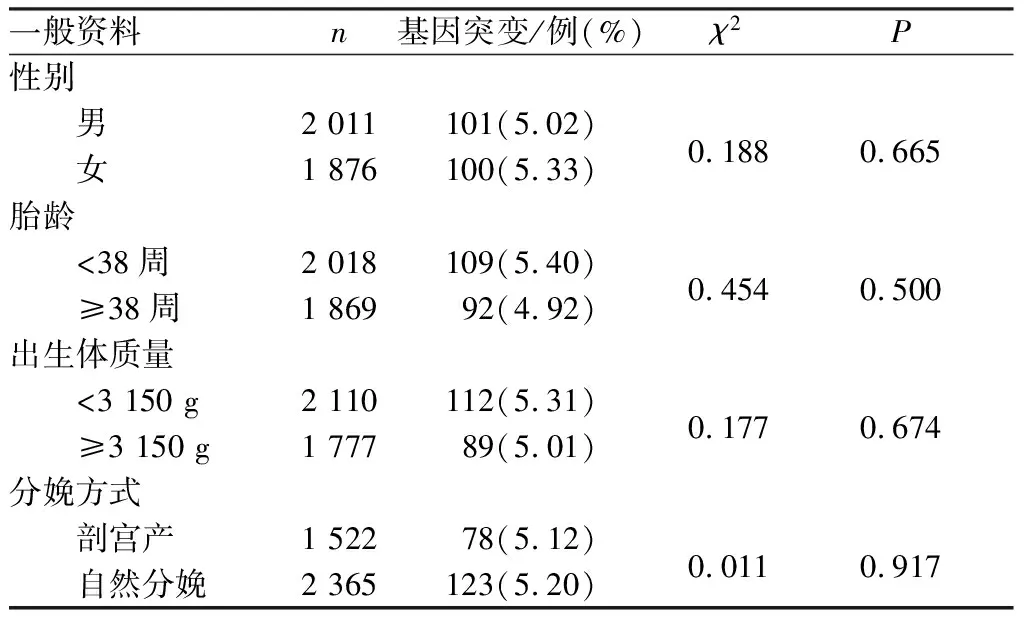
表2 不同性别、胎龄、出生体质量及分娩方式新生儿耳聋基因突变率比较
2.3 听力障碍发生情况结果见表3。3 887例新生儿中,经听力学评估最终确诊听力障碍28例,听力障碍发生率为0.72%(28/3 887);不同性别、胎龄、出生体质量及分娩方式新生儿听力障碍发生率比较差异均无统计学意义(P>0.05)。
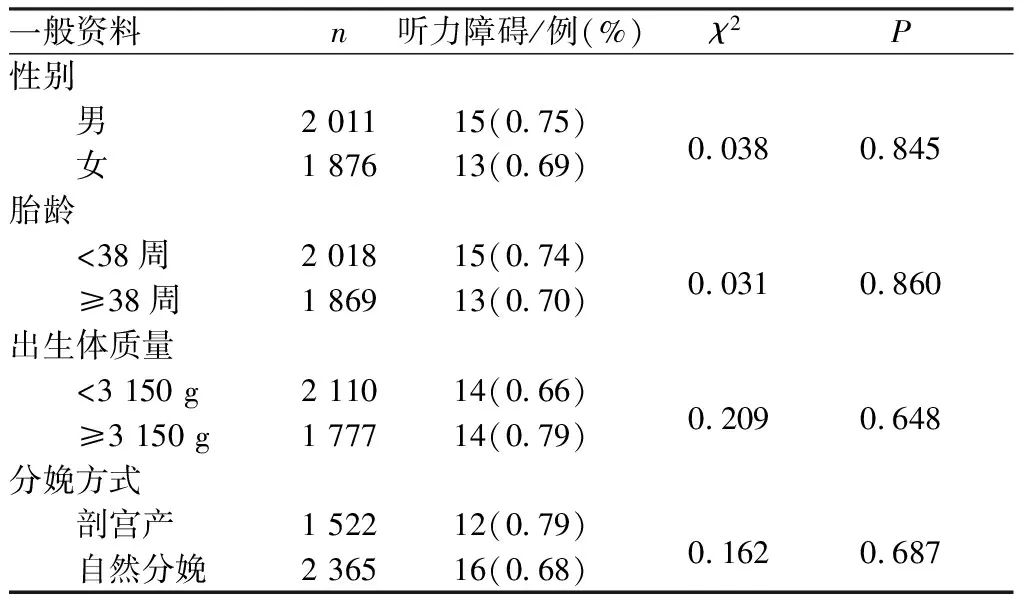
表3 不同性别、胎龄、出生体质量及分娩方式新生儿听力障碍发生率比较
2.4 耳聋基因突变与无基因突变新生儿听力障碍发生率比较201例耳聋基因突变新生儿发生听力障碍7例,听力障碍发生率为3.48%(7/201);3 686例无耳聋基因突变新生儿听力障碍21例,听力障碍发生率为0.57%(21/3 686);耳聋基因突变新生儿听力障碍发生率显著高于无耳聋基因突变新生儿,差异有统计学意义(χ2=18.724,P<0.05)。
3 讨论
听力检查是诊断新生儿听力障碍的主要方法,但是该方法尚存在一定的技术缺陷,例如对于药物敏感性耳聋及大前庭水管综合征不敏感,漏诊率较高[6-7]。另外,在大量的迟发性听力下降患儿中,亦有患者因自身基因缺陷致病,或由于基因缺陷和基因多态性造成对致聋环境因素易感性增加而致病[8-10]。
遗传学研究发现,人类基因中有200余个基因与耳聋有关,耳聋基因诊断可以筛选基因突变的个体携带者,在缺少有关耳聋家族史的情况下能对耳聋个体及其亲属做出相应智能基因的诊断,从而指导优生优育,尽可能避免同类耳聋的继续发生[9,11]。目前已公认Cx-26基因是耳聋较多见的致病基因,且其中75%~85%的遗传方式为常染色体隐性遗传,在此基础上可以运用适当的基因筛查方法和序列分析进行产前基因诊断,如胎儿确为此致病基因的携带者,可以规劝孕妇终止妊娠[11-14]。因此,遗传性耳聋的基因学诊断对于弥补常规听力检查的不足、提高诊断准确率及指导优生优育有重要意义。
本研究结果显示,3 887例新生儿中,筛查出耳聋基因突变者201例,耳聋基因突变率为5.17%;其中235delC位点杂合突变占40.30%,IVS7-2A>G位点杂合突变占25.87%,2168A>G位点杂合突变占12.44%,299-300delAT位点杂合突变占11.44%,其余位点突变率均低于10.00%;该结果提示耳聋基因突变具有一定的发生率,其中以235delC位点杂合突变和IVS7-2A>G位点杂合突变多见。235delC位点杂合突变属于GJB2基因。GJB2基因属于隐性基因,携带者并不发病,但与同样为携带者生育的后代听力障碍的发生率高达25.00%,因此,GJB2基因突变携带者需要定期检查和随访,并进行基因筛查,这对优婚优育具有一定的指导意义[15-17]。IVS7-2A>G位点杂合突变属于SLC26A4 基因突变,其中大前庭水管综合征属于SLC26A4 基因突变,正常条件下SLC26A4 基因突变患儿可能不发病,但如果出现感冒或头部撞击等极易导致听力损伤,因此,对于SLC26A4 基因突变的患儿需要提前加强护理和干预[18-19]。
本研究进一步对有耳聋基因突变与无耳聋基因突变新生儿听力障碍发生率进行了比较,发现耳聋基因突变新生儿听力障碍发生率为3.48%,无耳聋基因突变新生儿听力障碍发生率为0.57%,有耳聋基因突变新生儿听力障碍发生率显著高于无耳聋基因突变新生儿;该结果提示遗传性耳聋基因筛查有助于发现新生儿听力损伤并及时制定干预方案。常规的听力检查不仅需要相对安静的隔音室和配套的专业医疗设备,其检查结果也仅针对现阶段的听觉功能状态进行分析,对于未来可能出现的听力变化的新生儿筛查尚存在不足和局限性,因而结合基因筛查结果并采取有效的干预措施有助于患儿听力的保护和各种功能的正常发育[20]。针对已经确认存在听力障碍和耳聋的新生儿,在基因突变类型指导下进行针对性的功能训练有助于新生儿的健康成长。但是,由于耳聋致病因素复杂多样,基因筛查也可能存在一定的漏诊率,因此,临床需要结合听力筛查和遗传性耳聋基因筛查,提高诊断准确率。
综上所述,遗传性耳聋基因筛查有助于早期发现新生儿听力损伤,通过遗传性耳聋基因筛查能够筛查耳聋基因突变,依靠医学干预预防耳聋;第一时间筛查听力异常患儿,发现迟发性和药物性耳聋,有助于提高新生儿听力筛查的检出率;还可通过分子生物学水平对耳聋进行诊断,及早制定干预措施。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
中国民间疗法(2021年8期)2021-07-22 05:53:42
种子(2021年3期)2021-04-12 01:42:22
中国生殖健康(2020年4期)2021-01-18 02:58:32
中国生殖健康(2020年2期)2021-01-18 02:51:26
中国生殖健康(2018年4期)2018-11-06 07:12:36
小学生导刊(2018年13期)2018-06-29 03:49:00
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
中国中医药现代远程教育(2014年13期)2014-03-01 04:26:52
河南医学研究(2014年5期)2014-02-27 14:52:41
