
人工设计逆醛缩酶RA95.5-8F催化β-羟基酮C—C裂解的理论研究
2021-07-11 16:32李心怡刘永军
高等学校化学学报 2021年7期
李心怡,刘永军
(山东大学化学与化工学院,济南250100)
醛缩酶可以催化羟醛缩合反应,即亲核供体(可烯醇化的羰基,底物1)与亲电受体(通常是醛类,底物2)发生缩合形成C—C键[1].C—C键立体选择性的生成与断裂属于最基本的有机化学反应,因此醛缩酶在不对称合成中起十分重要的作用[2].目前,在自然界已发现了2类醛缩酶,其中Ⅰ类醛缩酶不需要辅因子[3,4],活性位点有一个保守的赖氨酸残基,ε-氨基可与供体底物的羰基反应并形成Schiff碱中间体[5],进一步转化成亲核性的烯胺形式;该烯胺再选择性地进攻醛受体的羰基,最后经过水解反应生成缩合产物.Ⅱ类醛缩酶需要二价金属离子(如Zn2+及Mg2+等)或辅酶作为辅因子,在反应中组氨酸首先抽取供体底物上的质子使底物形成烯醇负离子,然后对醛供体发起亲核进攻.绝大多数醛缩酶催化的羟醛缩合反应的立体选择性只受酶的控制,而不依赖于底物的立体构型[2].
迄今,一些有机胺和催化抗体等Ⅰ类醛缩酶的仿生催化剂已被合成出来[6,7],但其催化效率相对于天然酶还存在较大差距.近年来,随着计算模拟技术的发展,蛋白质分子设计技术已成为获得比天然蛋白质性能更优越的新型蛋白质的有力手段[8].Baker等[9,10]开发了一种用于蛋白质结构预测的Rosetta算法平台,并相继设计了很多具有重要生物功能的新酶,它们可以催化多种类型的化学反应,包括羟醛缩合反应[11~15]、Kemp消除反应(Kemp elimination reaction)[16]及狄尔斯-阿尔德反应等[17].2012年,Althoff等[12]设计了一种逆醛缩酶,可以催化羟醛缩合反应的逆反应,即β-羟基酮化合物的裂解反应(Scheme 1).2016年,Obexer等[18]利用超高通量液滴微流控技术对该逆醛缩酶进行了定向进化,获得了酶活性提升109倍的逆醛缩酶RA95.5-8F,尤其是通过6轮的突变和筛选后,RA95.5-8F的催化效率比起始进化酶RA95.5-8提高了30倍,且RA95.5-8F对R型和S型底物的立体选择性从14∶1提高到了480∶1,成为人工设计酶最为成功的案例之一.
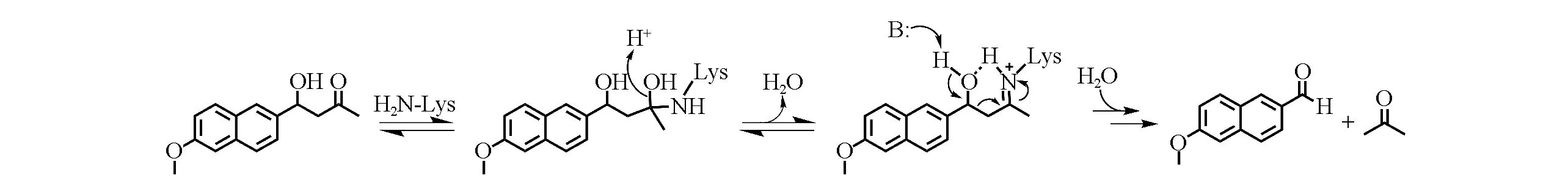
Scheme 1 Catalytic reaction catalyzed by retro aldolase
为了说明逆醛缩酶RA95.5-8F的高催化活性和对底物的立体选择性,Obexer等[18]还获得了高分辨率的RA95.5-8F酶的晶体结构,其活性中心包含一个由氢键网络连接的Lys1083,Tyr1051,Asn1110和Tyr1180组成的催化四联体[18~21](Scheme 2),这可能是影响酶催化活性和底物选择性的关键因素.在Obexer等提出的反应机理中,残基Lys1083首先亲核进攻供体底物羰基形成Schiff碱中间体,且四联体中的Tyr1051主要通过抽取底物上的质子起催化作用.为了进一步理解逆醛缩酶RA95.5-8F具有高催化活性的根本原因,本文采用QM/MM组合方法对逆醛缩酶RA95.5-8F的催化机理进行了理论研究,在原子水平上阐明了β-羟基酮化合物裂解的微观机制,解释了催化四联体中各残基所起的不同作用,研究结果为相关醛缩酶的进一步改造提供了理论参考.

Scheme 2 Crystal structures of the engineered retroaldolase RA95.5-8F with a bound 1,3-di ketone inhibitor(PDB ID:5AN7)(A)and active site of the retro-aldolase RA95.5-8F in which the inhibitor has been manually modified to the reaction intermediate(B)
1 计算方法与模型
1.1 模型构建
由于Obexer等[18]解析出的RA95.5-8F酶的晶体结构是酶与酰胺中间体形成的复合物(PDB ID:5AN7),本文首先将酰胺中间体上的羰基手动改为羟基构建了计算模型[Scheme 2(B)].羟基H原子有2种可能取向,本文根据其可与附近水分子形成氢键的特点确定了其优势构象.在此模型中,残基Lys1083已经与底物形成Schiff碱中间体,故在QM/MM计算中,以此反应中间体为起始点,分别研究Schiff碱中间体的形成及其进一步裂解过程.由于RA95.5-8F酶晶体结构缺失了残基Ser1058-Asp1061,本文利用Modeller软件包[22]将缺失的残基补全,并通过分子动力学模拟获得了较为稳定的计算模型.
1.2 分子动力学模拟
首先利用PROPKA程序[23]计算出实验pH值下可滴定残基的pKa值,并结合可视化的VMD程序[24]确定了各残基的质子化状态.根据Obexer等提出的可能反应机理[18],将活性中心的Tyr1051残基设为去质子化状态,而Tyr1180残基为质子化状态;体系中所有缺失的氢原子用CHARMM软件包[25]中的HBUILD模块进行添加.首先将整个蛋白质体系溶解在一个直径为3 nm的TIP3P[26]类型的水球中.为了保证体系的电中性,还随机向体系中加入了9个Na+,最终整个体系含有12386个原子,包括2761个TIP3P水分子.在进行分子动力学模拟之前,首先在CHARMM22/CMAP全原子力场[27]下对体系进行能量最小化,然后进行35 ns的分子动力学模拟.分子动力学模拟得到蛋白质骨架原子的RMSD变化如图1所示,可见约在7 ns后蛋白质骨架基本达到平衡.
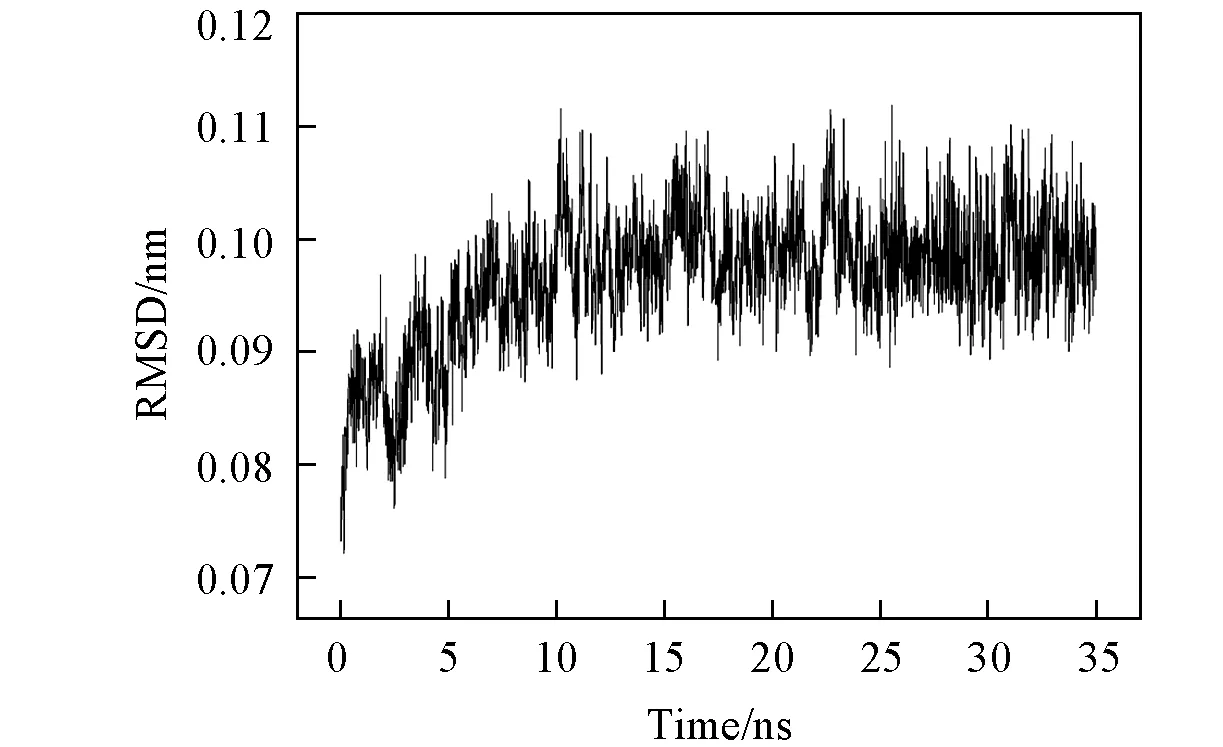
Fig.1 RMSD for the backbone atoms of the enzyme in complex with a Schiff base intermediate in 35 ns MD simulation
1.3 QM/MM计算
选取分子动力学模拟后的最后一个快照作为QM/MM计算的初始构型.所有QM/MM计算均使用Chemshell软 件 包[28],调 用Turbomole程 序[29]和DL_POLY程序[30]分别用于QM计算和MM计算.在计算中将整个体系分为QM区和MM区,其中QM区包括底物、参与反应的残基Lys1083、残基Tyr1051及与之形成氢键网络的残基Asn1110,Tyr1180和1个水分子(图2).QM区共有84个原子,电荷为0,自旋多重度为1.QM区与MM区的边界采用连接氢原子方法饱和[31].在QM/MM计算中,利用HDLC程序[32]进行几何构型优化,用L-BFGS方法[33,34]寻找反应路径中的能量最小点,采用P-RFO算法[35]寻找过渡态结构;所有几何优化均在B3LYP/6-31G(d,p)水平上进行[36].为了得到更准确的相对能量,对优化后的构型在B3LYP/6-311++G(2d,2p)水平上进行了单点能计算.此外,还用DFT-D3程序[37]对QM区进行了经验色散校正.

Fig.2 Selected QM region for the QM/MM calculations
2 结果与讨论
尽管本文进行的计算以Schiff碱中间体IM2为起始点,但为了清楚地描述β-羟基酮在RA95.5-8F酶催化下发生裂解的微观机制,仍然从底物β-羟基酮与残基Lys1083形成共价中间体开始说明.
2.1 烯胺中间体的形成与C—C键断裂

Fig.3 Optimized structures of species involved in the formation of the aldehyde and enamine
Obexer等[18]提出β-羟基酮的裂解反应是由Lys1083亲核进攻底物上的羰基碳引发的,本文首先对共价烯胺中间体的形成机制进行了研究.在优化得到的反应物构型中(图3),Lys1083氨基上的N1原子距底物C1原子仅为0.324 nm,非常有利于反应.此外,底物与附近残基形成多个较强的氢键,如Tyr1051的H2与底物O3相距0.168 nm,Tyr1180的酚羟基与底物羰基相距0.182 nm;Lys1083和Asn1110的氨基也分别与底物和Tyr1180存在氢键作用,极性活性口袋与底物形成了强大的氢键网络,从而使底物更准确地定位在酶的活性中心.计算结果表明,Lys1083亲核进攻底物羰基的同时,与N1相连的质子H1转移到O1原子上,形成一个以C1为手性碳的四面体结构IM1,反应能垒为77.82 kJ/mol.对于接下来的脱水形成烯胺过程,存在2种可能性,即底物上的羟基可与Tyr1051的H2或Tyr1180的H4结合形成水分子.IM1中O1与H2原子距离为0.172 nm,O1与H4原子的距离为0.197 nm,∠H1—O1—H2和∠H1—O1—H4分别为115.9°和94.1°.从相对距离和角度判断,羟基似乎更易与H2作用,计算结果也证实了这一点.羟基与H2结合脱水能垒为20.92 kJ/mol,而羟基与H4结合脱水的能垒略高,为25.10 kJ/mol.从IM1到IM2,随着底物OH-的离去,N1与C1形成烯胺双键,N1—C1键长也由0.142 nm缩短到0.131 nm.然后,去质子化的Tyr1051抽取底物上的质子(H3)促进底物C—C键的裂解.由于IM2中O2与H3相距较远(0.444 nm)且相对取向不利于反应,因此羟基需要先经历翻转.计算结果表明,羟基旋转过程十分容易,旋转后的中间体IM3也比旋转前更加稳定(图4).
烯胺中间体IM3的裂解(IM3→IM4)涉及多个化学键的断裂与形成,包括Tyr1051抽取底物上的质子和C2—C3键的断裂.计算结果表明,IM3→IM4过程的能垒为67.36 kJ/mol.底物的裂解首先导致产物醛的生成,同时生成另一分子的烯胺.C2与C3相距0.308 nm,说明醛碳基仍与残基Tyr1051形成氢键.由图4还可以看出,第一步Lys1083的亲核进攻过程对应的能垒较高,而脱水、羟基旋转等过程都很容易发生.

Fig.4 Potential energy profile for the formation of the aldehyde and enamine
2.2 烯胺水解反应及丙酮的生成

Fig.5 Optimized structures of species involved in the hydrolysis of the enamine and the release of acetone
图5给出烯胺水解及丙酮生成过程中优化得到的中间体和过渡态结构.可见,活性中心的水分子起到质子桥作用,Tyr1051上的质子首先传递水分子,同时水合质子加成到C2原子上生成IM5,由于C1=C2双键变为单键,其键长由IM4中的0.135 nm经过TS5(0.142 nm)拉长至IM5的0.148 nm,同时C1=N1双键形成,其键长由0.136 nm缩短至0.130 nm,此过程的能垒为72.38 kJ/mol.随后水分子的O—H键异裂,其中OH-亲核进攻C1原子,H+转移回Tyr1051上,生成IM6,此过程的能垒仅为25.52 kJ/mol(图6),说明烯胺水解反应比较容易发生.
决速步骤是丙酮分子的生成,中间体IM6需要经历C1—N1键的断裂及羟基O—H键的异裂,完成整个催化循环,此步的能垒为106.27 kJ/mol(图6).值得注意的是此过程与R→IM1的赖氨酸亲核进攻互为逆反应,因此二者能垒比较相近.此外,本文中的底物是R构型的β-羟基酮,在优化得到的反应物、中间体及过渡态构型中,底物的β-羟基始终与残基Tyr1051和Tyr1180等形成较强的氢键,有利于底物C—C键裂解反应的发生,这可能是导致此酶对于R构型底物的选择性远远高于S构型的原因之一.

Fig.6 Potential energy profile for the hydrolysis of the enamine and the release of acetone
2.3 残基突变计算
为了探讨由Lys1083,Tyr1051,Asn1110和Tyr1180构成的催化四联体对反应的影响,Obexer等[18]对四联体中的关键残基进行了突变,即将Tyr1051和Tyr1180突变为苯丙氨酸,将Asn1110突变为丝氨酸.基于此,本文也对上述残基突变进行了计算研究.优化后反应物的结构(图7)表明,底物不再被附近残基形成的氢键网络所稳定,但底物的羰基C1原子与Lys1083的N1原子的距离(0.320 nm)与突变前类似.本文计算了Lys1083对底物羰基的亲核进攻过程,结果表明,这一过程的反应能垒为157.23 kJ/mol,比突变前的能垒高79.41 kJ/mol,并且生成的四面体中间体IM1′比R′高34.12 kJ/mol.我们认为,反应能垒的升高可能是由于过渡态结构中缺少了底物与周围残基之间氢键的作用.此结果也说明,催化四联体对于稳定底物、降低反应能垒起到关键作用,这与Obexer等提出的猜想[18]相吻合.
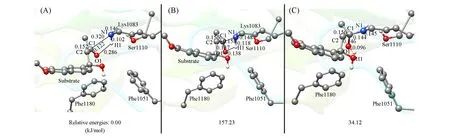
Fig.7 Optimized structures of R′to IM1′after the mutation of Tyr1051Phe,Asn1110Ser and Tyr1180Phe
基于计算结果,推测了逆醛缩酶RA95.5-8F催化β-羟基酮C—C裂解的详细机制(图8):赖氨酸首先通过亲核进攻底物羰基引发反应,并通过脱水生成Schiff碱中间体;在底物C—C键裂解过程中,去质子化的Tyr1051起关键的催化作用;在随后的水解反应中,活性位点的水分子起着质子桥的作用,质子化的残基Tyr1051通过此水分子桥将质子传递给底物,导致底物C=N双键中π键的断裂;最后,随着C—N键的断裂,底物羟基发生异裂,质子返回到赖氨酸上,生成丙酮分子.

Fig.8 Suggested catalytic mechanism of the retro-aldolase RA95.5-8F based on the QM/MMcalculations
3 结 论
利用QM/MM方法对人工逆醛缩酶RA95.5-8F催化β-羟基酮化合物的裂解反应进行了理论研究,结果表明,整个催化过程中最后一步C—N键的裂解是反应的决速步骤,能垒为106.27 kJ/mol.在催化循环中,由Tyr1180,Tyr1051,Lys1083和Asn1110组成的催化四联体起关键作用,Tyr1180和Tyr1051主要用于稳定底物在活性位点的位置与朝向,并通过与活性位点的水分子形成氢键在脱水、水解过程中发挥作用,而Asn1110则主要通过与Lys1083形成氢键来影响其与底物的结合,Lys1083和Tyr1051则分别作为亲核试剂和催化酸碱直接参与了催化反应.此外,催化四联体的氢键网络使得底物羟基更容易与R构型的底物结合,从而导致了RA95.5-8F对R构型底物的高选择性,这些研究结果可为进一步改进逆醛缩酶提供理论参考.
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
云南化工(2021年9期)2021-12-21
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
池州学院学报(2015年3期)2016-01-05
海南师范大学学报(自然科学版)(2015年2期)2015-12-23
天津科技大学学报(2015年2期)2015-08-09
重庆三峡学院学报(2014年3期)2014-07-16
