
电极材料储锂行为及其机制的原位透射电镜研究进展
2021-07-10 07:07柯承志肖本胜陆敬予张桥保
储能科学与技术 2021年4期
柯承志,肖本胜,李 苗,陆敬予,何 洋,张 力,张桥保
(1厦门大学材料学院,2厦门大学化学化工学院,福建厦门 361005;3哈尔滨工业大学(深圳)理学院,广东深圳 518055;4北京科技大学,北京市材料基因工程高精尖创新中心,北京 100083)
锂离子电池因具有能量密度/功率密度高、循环寿命长等优势在全球能源存储技术中占据了日益重要的地位[1-4]。然而,便携式电子设备和电动汽车及大型电网储能设备的快速发展,对下一代锂离子电池能量密度和功率密度提出了更高的要求。按照我国《节能与新能源汽车技术路线图》,2030年目标为500 W·h/kg。当前采用三元正极材料和石墨负极材料的锂离子电池,能量密度极限在250 W·h/kg左右[5-7]。先进电极材料开发是实现高能量密度锂离子电池的关键前提,因此寻求和开发新型高性能电极材料是研制下一代高能量密度锂离子电池发展的迫切需要[8-12]。目前,由高比容量电极材料组配的高能量密度锂离子电池的研发方兴未艾,并获得了一些重要研究进展。然而,这些电池内部的高比容量电极在充放电过程中仍存在诸多问题,极大制约了其进一步应用。以负极材料为例:锂离子在高比容量负极材料中的反复嵌/脱,易导致其微观形貌、晶体结构和体积发生明显变化,并影响多个尺度的界面稳定性,极大程度制约了电池的电化学性能[13-15]。对于正极材料,锂离子在脱/嵌过程中,可能会造成正极材料局域晶格的扭曲、阳离子混占位、堆垛层错、相变、相分离、产生空穴与裂纹等情况,这些结构演变常伴随着电荷转移;涉及的电荷补偿机制和材料电子结构的演化,也将直接影响电池的电化学性能[16-17]。因此,从本质上理解电池在工况状态下、实际电化学过程中电极材料的形貌、成分和微观结构演变,揭示电极材料微观结构特征及其动态演化与电池宏观电化学性能间的构效关系,对寻找并更有效地开发性能优异的电极材料、深入理解电池的失效机理和优化电池性能至关重要,这些都非常依赖清晰、准确的先进原位表征手段[18-22]。
迄今为止,各种先进的原位表征技术,如原位扫描电镜、原子力显微镜技术、X射线光电子能谱技术、X射线衍射技术、拉曼光谱技术和核磁共振谱学技术等,被应用于直接监测电极材料在工况下电化学循环过程中的微观结构、电子结构、成分和物相的动态演变过程,为体相电极材料的研究提供了重要的相、成分转变过程和材料失效机制等信息[23-26]。然而在真实的嵌/脱锂条件下,电极材料的结构和化学演变一般发生在特定的纳米甚至原子尺度的局域区域,例如缺陷,电极材料的固/固界面,以及电解质和电极材料形成的固/固界面等[27]。上述表征技术受限于其空间或时间分辨率,无法提供电极材料在充放电过程中表/界面处的原子级结构和成分的动态演变信息[28-30]。原位透射电镜(TEM)具有优异的空间和时间分辨率,同时可加装能量色散X 射线谱(EDS)、电子能量损失谱(EELS)、选区电子衍射(SAED)、扫描隧道显微镜(STM)、原子力显微镜(AFM)等[31],可实现从纳米甚至原子层面实时、动态监测电极材料在工况下的微观结构演化、反应动力学、相变、化学变化、机械应力以及表/界面处的原子级结构和成分演化等,对电极材料微观动态演变行为和反应机理等进行精确表述,从而为高性能电极材料的构筑与性能调控提供微观依据和创新思路[32-33]。鉴于此,本文归纳了当前应用原位TEM 技术解析锂离子电池关键电极材料在充放电过程中的微观动态演变规律与失效机制的重要研究进展,包括多种正极材料和高比容量负极材料的原位TEM 研究,尤其是它们在电化学过程中微观结构、化学成分与物相动态演变等信息。此外,本文对原位TEM 表征技术当前存在的问题,以及借助原位TEM 技术研究二次电池的未来发展方向进行了展望和思考。
1 锂离子电池正极材料的原位TEM研究
自1991 年,日本索尼公司首次将锂离子电池商业化以来,锂离子电池已被广泛应用于手机、便携式电子产品和电动汽车。其中正极材料性能是制约其能量和功率密度进一步提升的主要瓶颈[34]。钴酸锂(LiCoO2,LCO)作为最早商业化的锂离子电池正极材料,具有电压高、压实密度高等优点,是一种较为理想的锂离子电池正极材料[35]。但是在商业应用中,其可供循环使用的实际容量只占其理论容量的一半多一点。研究表明,提升充电电压能够有效提升LCO 材料的容量,但同时会导致LCO 结构稳定性降低,影响电池的循环性能。然而,关于LCO在高充电电压下微观结构动态演变及失效机理仍需进一步深入研究且鲜有报道。这主要是因为当锂离子嵌入LCO 晶格时,仅产生微小的应变而不会引起LCO 材料的明显晶胞体积变化,进而难以被传统的表征技术甚至高分辨TEM技术观测到[36]。
2017年谷林研究员课题组[37]采用聚焦离子束切片法构建了以LCO 为正极,Y 与Ta 掺杂的LLZO(Li6.75La2.84Y0.16Zr1.75Ta0.25O12)为电解质,金(Au)为负极,可在球差校正TEM 中观察的纳米电池。作者采用先进的原位环状亮场(ABF)STEM 结合高角度环形暗场(HAADF)STEM成像技术在原子尺度下解析了LCO 在电化学过程中晶体结构演变过程。研究发现[图1(a)],LCO 在经过高压脱锂后,其初始的单晶相转变成了纳米尺寸的多晶,晶粒大小为5~15 nm。如图1(b)~(e)所示,其在[010]晶轴方向形成相干孪晶边界和反相畴边界晶界,且脱锂后的LCO 中只存在两种晶体取向。这主要是由于相干孪晶界界面能量相比反相域晶界能量较低,更容易在脱锂过程中形成。另外,受锂离子脱出以及可能的锂或钴离子在畴界累积影响,LCO 产生了(2.7±0.4)%的层间距膨胀。这可能是由于钴离子迁移至锂离子层间,诱发了LCO 晶格结构由层状向尖晶石和岩盐结构转变。此外,LCO的纳米结晶主要是因电极与电解质之间的点接触,使得脱锂时只有接触区域才能进行锂离子的运输,致使LCO 正极发生单晶-多晶态相转变。该工作首次采用球差校正STEM技术在原子尺度上实时监测了电化学脱锂过程中LCO 结构的动态演变过程和由单晶转变为由相干孪晶边界和反相畴边界相连的纳米尺度多晶体的现象,为理解LCO 高压失效机理提供了直观可靠的科学依据。
近年来,研究工作者采用非原位的表征技术[39]对LCO嵌锂后的产物也进行了深入研究,Shu等[40]研究表明LCO在过嵌锂后最终转化成Co金属,并提出3种反应中间产物分别为Li1+xCoO2−y、Co3O4和CoO。Weker 等[41]借助X 射线吸收光谱(XAS)结合透射X射线显微镜(TXM)研究了在嵌锂过程中LCO转变为核壳结构Li2O和Co金属的演变过程。然而,从纳米甚至原子尺度上直接观测LCO 在嵌锂过程中微观结构和相变演化过程却未见报道。针对上述问题,Yang 等[38]应用原位TEM 在纳米尺度下研究了嵌锂过程中LCO 的物相演变。作者利用稳定的惰性SrTiO3(STO)作为载体,采用脉冲激光沉积(PLD)的方法,在STO上沉积一层SrRuO3(SRO)缓冲层,最后将LCO 薄膜生长在SRO 缓冲层上,从而保证LCO 在电子束照射下结构的稳定。在不施加电压的条件下,当钨针尖上的锂金属电极与LCO接触时,即发生电化学反应。利用纳米束斑电子衍射(NBD)和电子能量损失能谱(EELS)相结合的方法来确定嵌锂前后形成的反应物。图1(f)中LCO/SRO 界面显示为LCO 的[001]晶面与SRO 的[001]晶面。图1(g)显示了LCO/SRO/STO与锂金属接触后的不同区域,其中标记1号的为原始状态的LCO区域。从图1(h)中的NBD 结果得出LCO 晶粒出现了相互叠加的现象。4 号标记部分为完全嵌锂后的区域,NBD 证实了其反应产物为Co 金属和Li2O[图1(k)]。因NBD 具有高时空分辨率的特点,可以观测到接近反应端的中间产物。在接近反应端的3 号标记区域,NBD 的结果显示了Co 金属和CoO共存的衍射斑点[图1(j)]。在点2的区域,NBD的结果证实了Co 金属相关的衍射斑点消失,同时CoO 相关的衍射斑强度增加[图1(i)]。完全嵌锂后的区域宽度为125 nm 与原始LCO 宽度(105 nm)相比膨胀了19%。该项研究表明Li 与LCO 正极直接接触会导致LCO 发生膨胀和不可逆相变,最终转变成Co金属和Li2O,其中CoO作为亚稳态反应中间相。该研究对深入理解LCO 在嵌锂过程中的失效机理和进一步改性LCO 的电化学性能具有重要的指导意义。
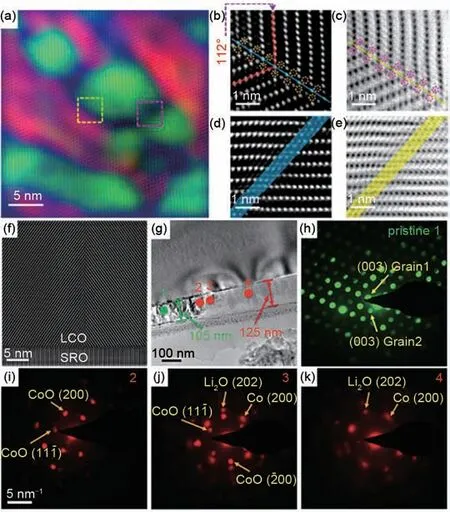
图1 (a)采用GPA方法着色后脱锂的LiCoO2正极HAADF图;(b)和(c)是黄色虚线矩形区域放大的原子级分辨TEM图;(d)和(e)是粉色虚线矩形区域放大的原子级分辨TEM图[37];(f)原始LCO薄膜样品的HAADF-STEM图;(g)嵌锂后的低倍TEM图;(h)~((k)在(g)中标记不同点的NBD图[38]Fig.1 (a)HAADF image of delithiated LiCoO2 cathode after coloring with GPA method,(b)and(c)are zoomed-in views of yellow,dashed-box area,(d)and(e)are zoomed-in images of pink,dashed-box area[37],(f)A HAADF-STEM image of a pristine thin film LCO piece,(g)A TEM image after lithiation acquired at a low magnification,(h)~(k)NBD patterns for different spots labeled in(g)[38]
橄榄石型磷酸铁锂(LiFePO4,LFP)因具有安全性能好、循环寿命长、原材料来源广泛、无环境污染等优点被认为是极具发展潜力的锂离子电池正极材料,在学术界和工艺界引起了广泛的关注和研究。然而其电子导电性差和锂离子扩散速率低等缺点也制约了锂离子电池性能的进一步提高。近年来,通过掺杂、表面碳包覆和纳米化的策略,LFP正极的电化学性能已得到了大幅提升。虽然各种非原位表征技术对LFP的电化学机理已有了较充分的认识,然而其在充放电过程中的微观结构演变和相转变及失效机制报道甚少。这主要是由于锂离子在LFP材料嵌脱过程中伴随的局域结构变化常发生在纳米甚至原子尺度上,而难以被常规的X射线等结构研究方法表征。这些局域结构的演变与电极的电化学性能密切相关,因此深入理解这些演变行为对于提高电池宏观性能至关重要。
Zhu 等[42]通过水热法合成了LFP 材料,采用化学脱锂法制备得到了FePO4。通过构建类似如图2(a)所示纳米电池成功地在原子尺度实时观察到了FePO4在嵌锂过程中通过相界迁移而不是通常认为的固溶体途径转变为LFP的过程[图2(b)~(i)]。如图2(c)所示,为了提高其电导率,作者在晶体FePO4表面包覆了一层10 nm 厚的非晶碳(α-C)。随着锂的嵌入,α-C 层和FePO4之间形成了较薄的LFP 层。嵌锂176 s 后,生成了较厚的LFP 层[图2(d)]。随着更多的锂离子嵌入到FePO4中,LFP的厚度从6.4 nm增加到11.3 nm,同时相界面(PB)从表面向核内迁移[图2(e)、(f)]。经过282 s的嵌锂后,在FePO4和LFP 之间的(020)晶面上形成台阶较小的PB,表明锂离子是从[010]方向插入到FePO4中[图2(g)~(i)]。LFP层厚度随着PB沿[010]方向按照阶梯状的方式传递而增加,PB 的移动台阶也相应沿[010]方向迁移。该研究实时、动态地解析了FePO4在电化学过程中的嵌锂机理,观测到了在LFP 和FePO4之间存在一个清晰的(010)相边界且在嵌锂过程中沿[010]方向迁移,与锂离子扩散方向相同。这也是首次从原子尺度上原位实时地研究了FePO4材料中相边界迁移和各向异性嵌锂的机制,为LFP在电化学过程中的嵌锂机理研究提供了科学依据。
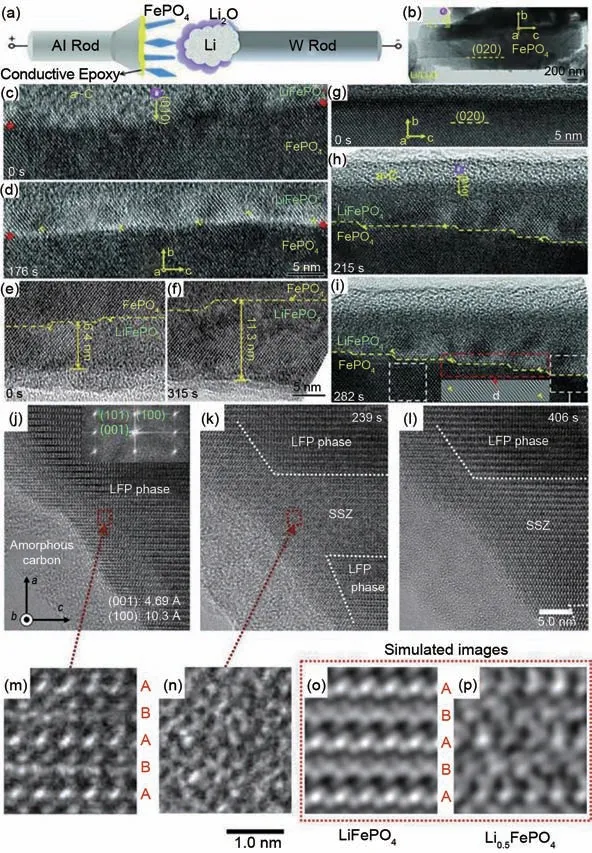
图2 (a)为原位TEM纳米电池示意图;(b)为FePO4和固态电解质Li/Li2O接触后的TEM图;(c)~((i)在嵌锂时FePO4随时间变化的TEM图[42];(j)~(l)为LFP纳米线在嵌锂过程中的微观结构演变图;(m)、(n)在(j)、(k)中红色虚线矩形图像的放大图;(o)和(p)为LFP和Li0.5FePO4的TEM模拟图[43]Fig.2 (a)Schematic diagram of in situ TEM setup with nanobattery,(b)A TEM image of FePO4 particle after its contact with Li/Li2O solid electrolyte,(c)~(i)Time-lapse TEM images of FePO4 particle during lithiation[42],(j)~(l)Microstructural evolution of a LFP nanowire during lithiation,(m)、(n)Magnified images of dotted red rectangle region in(j)、(k),respectively.Simulated TEM images of(o)LiFePO4 and(p)Li0.5FePO4[43]
随后,Niu 等[43]通过静电纺丝的方法合成了LFP 纳米线,利用原位TEM 研究了其脱锂过程的局部区域结构变化。图2(j)~(l)为LFP 纳米线在脱锂过程中的高分辨率TEM图,1.03 nm和0.469 nm的晶面间距分别对应于LFP 初始状态的[100]面和[001]面。当脱锂239 s后,如图2(k)所示,出现了较大的固溶体区(SSZ),面积约为11 nm×22 nm。与图2(m)初始状态的LFP 相比,图2(n)显示SSZ区域在脱锂过程中出现了明显的无序结构。进一步脱锂到406 s,如图2(l)所示,在无序SSZ的区域处再次出现了清晰的晶格条纹。图2(o)的LFP 模拟图能很好地匹配图2(m)初始状态的LFP 高分辨TEM 图,同时,Li0.5FePO4模拟图[图2(p)]与图2(n)中的无晶格条纹区也非常相似。该研究利用原位高分辨率TEM 首次观察到在脱锂过程中,锂离子固溶体区(Li0.5FePO4)在表面形成并迅速长大,这与他人报道脱锂过程中存在明显LFP/FePO4界面不同。而且与他人看到的界面晶格失配不同,在LFP/FePO4间存在的固溶体过渡区域界面没有位错,因此降低了界面断裂的可能性,这正好解释了该正极材料可获得长循环和高倍率的重要原因之一。该研究揭示出一些基本电化学问题中存在的材料结构和性能的密切关联,为今后更好地理解并设计高性能LFP正极材料提供了科学依据。
与LCO 和LFP 相比,高镍层状材料(LiNi1−x−yMnxCoyO2,NCM)因具有经济效益好和放电容量高的优点而备受关注,但较差的循环稳定性和倍率性能阻碍了其商业化应用。大量研究表明其电化学性能衰减跟材料表面的结构变化相关。然而这些研究很少考虑晶体结构缺陷,例如位错、堆叠层错、反相界面以及孪晶界与高镍正极电化学性能间的构效关系及作用机制。近日,中科院物理所苏东研究员课题组[44]在TEM 下构建了以LiNi0.76Mn0.14Co0.10O2(NCM76)为正极,Li0.5La0.5TiO3(LLTO)为电解质,钨针尖作为负极的纳米电池[图3(a)、(b)],实时动态地观测了NCM76 在脱锂过程中的电极材料内部由晶体缺陷诱导的结构演化现象[图3(c)~(h)]。研究发现在原位脱锂过程中,反向畴界(APB)处的不全位错移动导致畴界延伸,并且该现象表现出各向异性的特点。同时还发现在共格孪晶界(TB)处锂原子和过渡族金属原子存在明显的混排现象,且在该处首先发生层状结构向岩盐结构的转变。因其具有较小的扩散势垒,TB 可为过渡族金属原子和锂原子提供扩散通道。同时,可以促进阳离子无序及岩盐相的形成。从结构角度来看,因APB 的扩展阻碍了锂扩散的通道,致使锂离子脱/嵌的阻抗增加,并且岩盐相的形成使得电极在电化学循环过程中容量进一步衰减。该项研究首次观察到了颗粒内部由晶体缺陷诱导的第二相产生,证实了高镍NMC 材料的衰减机理并不是只由表面结构决定,其颗粒内部由缺陷导致的锂不均匀脱嵌和过渡族金属离子的迁移也会对性能产生影响,揭示了晶体结构缺陷与NMC材料性能衰减存在着直接的关系。
尖晶石结构的镍锰酸锂(LiNi0.5Mn1.5O4,LNMO)因其工作电压高、安全性能好、成本低廉、对环境友好等优点,是极具前景的高能量密度锂离子电池正极材料。然而其在商业化应用中仍存在着一些挑战,LNMO在充电过程中表面形成的高浓度、强氧化性的Mn4+和Ni4+,会加剧电极表/界面副反应,影响着材料的电化学性能。针对这些问题,需要深入理解LNMO在充放电过程中的微观结构演变和失效机制,以期进一步改善LNMO 的电化学性能。近期,谷林研究员团队[45]借助原位球差校正TEM在原子尺度下实时动态观测了P4332空间群立方相尖晶石LNMO在脱锂过程中电子结构的演变过程。研究发现在LNMO 正极材料中由于固/固界面的不均匀接触导致电极材料脱锂不均匀现象[图4(a)~(h)],进而造成局部区域出现过渡金属堵塞了锂离子输运通道。为了更好地从三维角度研究LNMO原子尺度下的结构演变,作者沿[111]、[110]和[100]轴对LNMO 进行了原位脱锂实验。如图4(i)~(k)所示,在脱锂过中,LNMO的结构发生了从有序到无序的演变。研究表明,在脱锂过程中,LNMO 在沿轴[112]的方向上形成了过渡金属富集区、反相边界区和过渡金属迁移前缘区,这主要是由锂离子的不均匀脱出导致过渡金属离子的局部迁移和反相边界的形成引起。不同于沿轴[112]方向的非均匀迁移,锂离子在沿轴[100]、[110]、[111]方向都为均匀迁移,LNMO表现出由有序结构向无序结构的转变,使原本有序的Ni/Mn结构变成完全的无序。该项研究不仅表明原子尺度下的三维表征对于提高对LNMO脱锂的动力学过程和基本电化学机理的理解的重要性,而且还为进一步优化LNMO结构稳定性提供了强有力的指导。

图3 (a)为原位TEM纳米电池图;(b)为低倍HAADF-STEM图;(c),(d)为脱锂前后NMC的HR-HAADF-STEM图;(e)为相扩展和缺陷迁移的TEM图;(f)~(h)为(c),(d)中的放大图[44]Fig.3 (a)Schematic of in situ TEM experimental setup;(b)low magnification HAADF-STEM images;(c),(d)HR-HAADF-STEM images of NMC before and after delithiation,respectively;(e)Phases extension and defects migration;(f)~(h)Magnified images of(c),(d)[44]
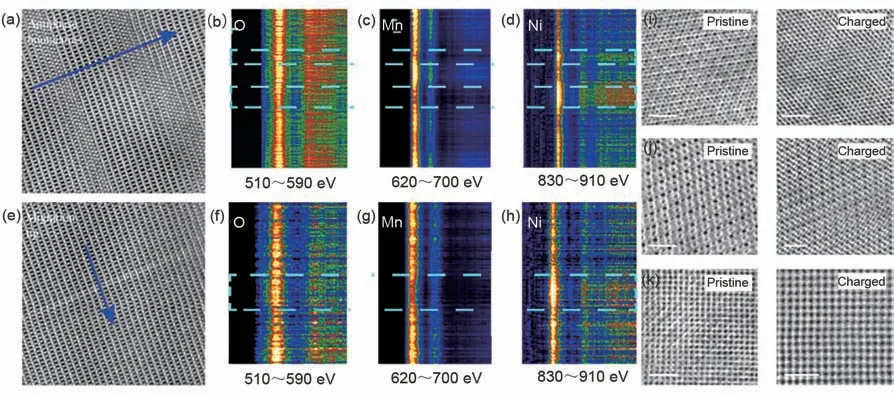
图4 (a)为反相边界区的HAADF-STEM图;(b)~(d)为在反相边界区中的O、Mn、Ni的EELS谱图;(e)为迁移前缘区的HAADF-STEM图;(f)~(h)为在迁移前缘区中的O、Mn、Ni的EELS谱图;(i)~(k)为原始和充电后LNMO沿[111]、[110]和[100]晶向的ABF-STEM图[45]Fig.4 (a)HAADF-STEM image of antiphase boundaries,(b)~(d)EELS spectrum of O,Mn,and Ni from(a),respectively,(e)HAADF-STEM image of migration-front area,(f)~(h)EELS spectrum of O,Mn,and Ni from(e),(i)~(k)ABF-STEM images of pristine and charged LNMO along[111],[110]and[100]direction,respectively[45]
2 锂离子电池负极材料的原位TEM研究
负极材料作为锂离子电池的重要组成部分,亦是影响锂离子电池能量密度和循环寿命的重要因素之一。根据负极材料储锂的机理,可以将其主要分为嵌入型负极、转化型负极和合金化型负极三类。相比现有嵌入型石墨负极材料,合金化型(如硅、锡、锑等)和转化型(如过渡金属氧化物、硫化物和磷化物等)负极材料具有更高的理论比容量,在提高能量密度方面具有优势,因此是下一代高能量密度锂离子电池备受关注的两类负极材料。然而,这两类高比容量负极材料在嵌脱锂过程中普遍存在物相转变及体积变化大的问题,导致首次充放电不可逆容量高和循环过程中容量快速衰减致使其电化学性能欠佳。利用原位TEM 技术深入研究这些高比容量负极材料在电化学过程中的微观结构、形貌、物相和化学组成的动态演化对电池性能的影响及作用机制,对优化和设计高比容量负极材料具有重要的指导意义。本文将针对部分相关核心工作进行详细评述[46]。
2.1 合金化型负极材料
合金化型储锂负极材料最典型的代表就是Si负极,由于其超高理论比容量(4200 mA·h/g)且其嵌/脱锂电位适中、在地壳中储量丰富等优点,已被广泛研究并被认为是下一代锂离子电池负极材料的理想选择[47-49]。大量的研究表明导电性差、嵌脱锂过程中巨大的体积变化(>300%)和固态电解质界面膜(SEI)始终处于破坏-重构的动态变化是导致Si负极倍率和循环性能欠佳的主要原因[50-52]。为了更有效地设计高性能Si基负极材料,需对其在工况下的基本物理化学性质(微观形貌、晶体结构、化学组成)及其动态演化对电池电化学性能的影响和作用机制进行深入探讨,以期为解决目前Si基负极中存在的瓶颈问题提供新的研究思路和重要科学依据[53-54]。原位TEM 技术为深入理解Si 负极在充放电过程中的动态过程和失效机制提供了直接证据。
Lee等[55]采用原位TEM研究了不同晶面取向的晶体Si纳米颗粒在第1次嵌锂时的体积和形状的动态演变过程[图5(a)~(l)],发现了Si 的各向异性膨胀的现象。研究证实,具有[100]、[110]和[111]取向的晶体Si 纳米颗粒在嵌锂过程中形成的非晶相(LixSi)首先在Li+浓度最高的[110]晶面的表面形成,形状也从最初圆形截面分别扩展成交叉、椭圆和六角形。图5(m)和(n)统计了3 种类型Si 纳米颗粒的截面尺寸变化和面积变化数据。在嵌锂的初始阶段,Li+沿[110]晶面进入晶体Si纳米结构中,并通过断裂Si-Si 键诱导[111]取向塌陷,致使产生[111]和[100]取向的纳米颗粒高度收缩,而[110]取向的纳米颗粒高度增加的各向异性膨胀现象。同年,Liu等[56]通过原位TEM实时观测了Si纳米线在嵌锂过程中的微观结构演变,也证实了Si 的各向异性膨胀现象。研究发现,锂化反应首先发生在Li 和Si 接触的地方,并沿着纳米线的轴线逐渐进行。由于Si 纳米线外层的锂化速度比内部的锂化速度要快,致使中心是未反应结晶态的Si(c-Si),而外层是已锂化完全的Li15Si4合金相。随着嵌锂的进行,中心未反应的c-Si呈渐缩状,产生裂纹并随之扩展。同时伴随着中心c-Si的完全消耗,裂纹扩展停止[图5(o)]。最后,由于塑性流动和锂化壳内的拉伸环向应力累积引起的不稳定,各向异性的体积膨胀使Si 纳米线产生了哑铃状截面[图5(p)]。Li 扩散耦合弹塑性变形模拟[图5(q)]进一步证实了嵌锂过程中Si 纳米线的各向异性膨胀和破裂现象。
为了解释Si 纳米线各向异性膨胀的本质原因,Liu 等[57]借助高分辨TEM 从原子尺度上揭示了单晶Si(c-Si)纳米线的嵌锂动态结构演化过程,发现了嵌锂过程中Si 纳米线的体积膨胀主要发生在[110]方向,而沿[111]方向几乎没有体积膨胀。如图6(a)~(d)所示,Si纳米线沿[111]取向嵌锂,随着嵌锂的进行,在c-Si外层形成非晶态的a-LixSi合金层。原位高分辨TEM证实[图6(e)~(g)],在c-Si和非晶态LixSi 合金之间存在一个锐利界面(ACI),此界面只有1 nm厚。此外,ACI的迁移主要沿c-Si纳米线[111]晶面的横向移动,即非晶态LixSi 合金是通过Si[111]原子平面逐层剥离而产生的[图6(h)~(j)],证实嵌锂过程是由短程的界面移动控制,而不是常规的扩散控制。进一步研究表明,在[110]面上有许多[111]台阶,而这些台阶正好是锂进入晶体Si的通道;但[111]面上却没有这样的台阶或嵌锂通道。由于Li+仅在致密Si[111]面的[110]和[112]方向上嵌入c-Si,且ACI在[111]面的迁移比在[110]和[112]面的迁移速率至少慢一个数量级,这导致界面流动具有很强的取向依赖性。因此这种各向异性的界面迁移导致Si纳米线在不同方向上产生截然不同的膨胀,揭示了Si产生各向异性体积膨胀的本质原因。
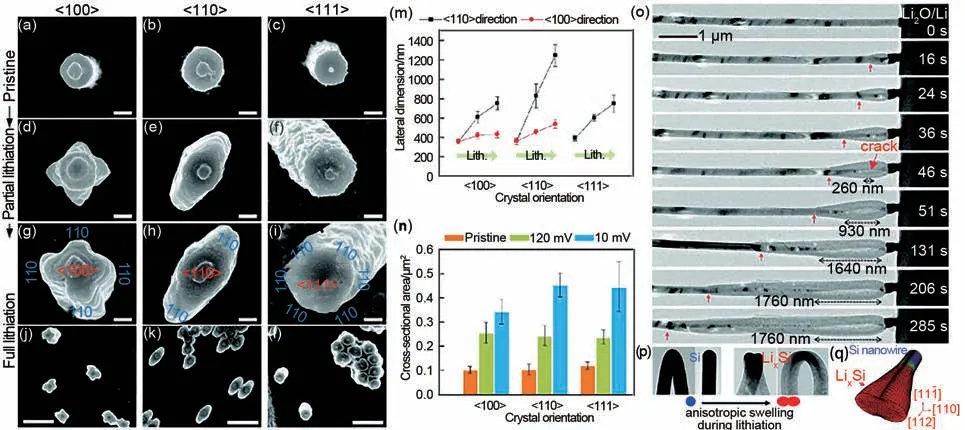
图5 (a)~(l)Si纳米柱沿[100]、[110]及[111]晶向在不同锂化状态下的SEM顶视图;(m),(n)Si纳米柱各晶向对应横截面尺寸和横截面面积变化的统计数据图[55];(o),(p)Si纳米线在锂化过程中微结构演变的TEM图;(q)逐步锂化的Si纳米线的三维模拟图[56]Fig.5 (a)~(l)Top-view SEM images of Si nanopillars along[100],[110]and[111]directions at different lithiation states;(m),(n)Statistical data of changes in cross-sectional dimensions and cross-sectional area along these three orientations[55];(o),(p)Microstructural evolution of Si nanowire during lithiation;(q)3D simulation plot of a progressively lithiated Si nanowire[56]

图6 (a)~(d)原始Si纳米线和部分锂化后Si纳米线的TEM图及对应的电子衍射图;(e)~(g)锂化过程中ACI随时间迁移的TEM图;(h)~(j)ACI的锂化过程,以及剥落过程的TEM图及对应示意图[57]Fig.6 (a)~(d)TEM images and corresponding electron diffraction patterns of a pristine silicon nanowire and a partially lithiated silicon nanowire;(e)~(g)Time-lapse images showing migration of ACI during lithiation;(h)~(j)TEM images and diagrams of lithiation process by lateral ledge flow in ACI and simultaneous peeling-off[57]
由于制约Si负极材料应用的最重要问题就是体积膨胀带来的断裂甚至粉化造成容量急速衰减。为了理解Si 纳米颗粒在嵌锂过程中的体积变化行为,Liu 等[58]采用原位TEM 技术实时动态研究了不同直径的Si 纳米颗粒的嵌锂行为,发现Si 纳米颗粒是否产生裂纹跟颗粒的尺寸相关,临界尺寸为150 nm[图7(k)]。研究发现当Si纳米颗粒的直径低于150 nm 时,Si 纳米颗粒即使发生巨大体积膨胀也不会有裂纹和破裂产生[图7(g)~(j)]。然而,当Si纳米颗粒的直径大于150 nm后[图7(a)~(e)],Si纳米粒子在嵌锂过程中首先在表面产生裂纹,并随着嵌锂的进行裂纹快速向内扩展。同时,在纳米颗粒不同位置出现新的裂纹,最终纳米颗粒由于嵌锂产生膨胀而破裂、粉化。电子衍射结果证实嵌锂后单晶纳米颗粒Si 转变成了多晶的Li15Si4合金[图7(f)]。此外,尺寸大于150 nm 的硅纳米颗粒的表面断裂行为的出现是由于嵌锂产生的拉伸应力超过了Si材料的屈服强度,驱动了裂纹扩展。这些结果直接证实了使用尺寸较小的Si纳米颗粒有利于提高锂离子电池循环过程中的结构稳定性,这为高性能Si基或其他合金化型负极材料的结构设计和性能优化提供了重要科学依据。
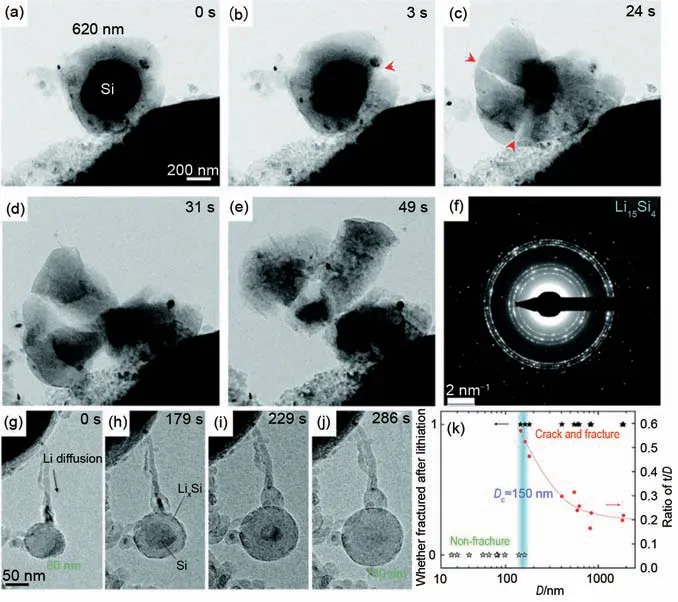
图7 (a)~(e) 620 nm Si纳米颗粒在嵌锂过程逐渐膨胀破裂的TEM图;(f) 嵌锂后的电子衍射图,对应的产物为Li15Si4;(g)~(j) 80 nm Si纳米颗粒在嵌锂过程中的结构变化图,锂化后没有破裂;(k) Si纳米颗粒嵌锂破裂的临界尺寸统计数据图[58]Fig.7 (a)~(e)TEM images showing fast fracture of 620 nm Si nanoparticle during lithiation process.(f)Electron diffraction pattern of broken Si nanoparticle after lithiation,indicating formation of Li15Si4.(g)~(j)TEM images of a 80 nm Si nanoparticle without cracking during lithiation.(k)Statistics showing critical size of Si nanoparticles without cracking during lithiation process[58]
近年来,研究工作通过对Si进行结构设计并结合有效包覆,已大幅改善了Si负极的循环稳定性[59]。然而,受制于表征手段的空间分辨能力,包覆层和结构设计对提升Si负极电化学性能的微观作用机制尚不清晰。Zhang等[60]创新性地构筑了三维多孔泡沫镍支撑的中空铜(Cu)线核外包覆Si和锗(Ge)双层壳结构[图8(a)~(c)],利用原位TEM技术首次实时研究了Cu/Si/Ge纳米线在嵌锂过程中形貌的动态演化过程[图8(d)],证实外层Si包覆可改变纳米线的嵌锂行为:其在嵌锂过程中轴向上发生了弯曲伸长(118%)和径向上向外膨胀(仅123.6%);这揭示了外层Ge包覆可有效缓解硅在径向上的巨大体积膨胀。此外,进一步实时监测了嵌锂过程中Si/Ge厚度随嵌锂时间变化的动态过程[图8(e)、(f)],首次发现复合纳米线的外层Ge先发生嵌锂,随后Si和Ge界面呈现“共嵌脱锂”的特殊新机制,且整体电极结构在反复嵌/脱锂过程中保持完整未发生破碎。这直接证明了Si、Ge间的相互协同作用可有效缓解Si体积膨胀问题,并显著提高电极结构稳定性。该项工作不但为设计和合成高性能锂离子电池用Si复合负极提供了新的思路,还为今后更好地理解Si复合负极材料中多个活性组分之间的协同作用反应机理打开了一扇崭新的窗户,从而为设计高性能锂离子电池负极材料提供指导。

图8 (a)Cu/Si/Ge纳米线的TEM图;(b)Si/Ge双层膜的HRTEM图及其SAED图;(c)Cu/Si/Ge NW的STEM图和相应的EDX谱图;(d),(e)Cu/Si/Ge纳米线随嵌锂时间变化的TEM图;(f)Si/Ge双层壳层厚度随时间变化测量图[60]Fig.8 (a)TEM image of a Cu/Si/Ge nanowire;(b)HRTEM image of Si/Ge bilayer and corresponding SAED pattern;(c)STEM image of Cu/Si/Ge nanowire and corresponding EDX spectra;(d),(e)Time-lapse TEM images of Cu/Si/Ge nanowire during lithiation;(f)Evolution of layer thickness in Si/Ge bilayer shell during lithiation[60]
对Si材料进行多孔化和表面碳包覆,也被认为能较好地解决其体积膨胀大和导电性差的问题,从而改善Si负极的电化学性能[61-64]。An等[65]构筑了三维纳米结构Si骨架组成的类蚁巢状多孔微米Si,并对其进行碳包覆形成类蚁巢状多孔Si/C 核壳结构,有效解决了嵌/脱锂过程中硅体积膨胀的问题,提高了电极结构稳定性。作者利用原位TEM 实时研究了类蚁巢状多孔微米Si/碳电极在嵌锂过程中微观结构和形貌的动态演化,直接证明了嵌锂过程中三维互连的硅纳米骨架能够有效地分散锂化过程中的应力并牵制向外的体积膨胀,内部三维贯穿的孔结构有效容纳硅纳米骨架向内的体积膨胀,电极结构嵌锂过程中体积变化小[图9(a)~(f)]。此外,原位TEM 多次循环研究发现类蚁巢状的Si 纳米骨架可在循环过程中可逆地膨胀/收缩而不粉化,并且Si的体积膨胀可通过周围的孔隙可逆地向内呼吸从而导致可忽略的颗粒水平外膨胀[图9(g)~(j)],这很好地解释了类蚁巢状多孔Si/C获得优异长循环稳定性的本质原因。该研究建立了材料微观结构与电化学性能间的构效关系,可为材料储锂性能、体积效应与材料多孔结构间关联规律的研究提供新的实验证据和重要的思路,为指导多孔材料结构设计及电化学性能优化提供重要科学依据。此外,Cui等[66]通过原位TEM 研究了具有空腔结构微米级Si@石墨烯在嵌/脱锂过程中的电化学行为,揭示了其获得优异循环稳定性的内在原因。研究发现Si材料外部包覆石墨烯可以同时有效提高界面电导率并缓解大体积膨胀带来的应力,另外二者之间的空隙给Si体积膨胀和收缩提供充足的空间,显著提升了复合负极的倍率和循环性能。

图9 (a)~(f)为AMPSi@C电极在嵌锂过程中的TEM图;AMPSi@C在第一次循环后(g),第二次循环后(h),第三次循环后(i)和第四次循环后(j)的原位TEM图[65]Fig.9 (a)~(f)Time-lapse TEM images of AMPSi@C electrode during lithiation.In situ TEM images of AMPSi@C(g)after 1st cycle,(h)after 2nd cycle,(i)after 3rd cycle and(j)after 4th cycle[65]
针对其他类型的合金型材料,Liu 等[67]通过原位TEM研究了Ge纳米线嵌脱锂行为。研究发现嵌锂后形成晶体Li15Ge4相,而脱锂后会形成由柯肯达尔效应导致高密度空洞形貌的非晶Ge 纳米线,如此循环往复,结构和空洞均可逆。同时,研究证实Ge 在嵌锂过程中发生的是各向同性的体积膨胀,这与同主族Si 的各向异性体积膨胀不同。Boebinger 等[68]采用原位TEM 监测了Sb 纳米颗粒在充放电过程中的微观结构、形貌和物相的演变过程。研究发现颗粒足够小的Sb 纳米颗粒在嵌锂的过程中发生膨胀并在脱锂过程中自发形成均匀的空腔且其可在循环过程中可逆地填充和形成空腔结构。此外,研究工作者借助原位TEM 技术也研究了Sn[69]、Ga[70]等合金负极材料在嵌/脱锂过程中的形貌、成分和微观结构变化,对深入理解合金负极失效机理和优化合金负极电化学性能具有重要指导意义。
2.2 转换型负极材料
转化型储锂负极材料,如过渡金属氧化物、硫化物和磷化物等,由于具有较高的理论储锂比容量也引起了研究者们的广泛关注[71]。对上述电极材料进行合理的结构设计和包覆改性,可显著改善其循环和倍率性能。但其在充放电过程中仍面临诸多挑战,例如:体积变化大,首次库仑效率低,电压极化大及不可逆的电化学反应等,且仍缺乏对其微观动态演化及失效机制的深入理解。因此,深入研究其在嵌脱锂过程中的动态演化过程与失效机制,对于更好地调控和优化它们的电化学性能至关重要。
作为一类重要的转化型负极材料,铁基氧化物(Fe2O3)因理论比容量高、储量丰富、环境友好且易于制备等优点,已成为国内外电化学储能领域的研究热点[72]。为了解决Fe2O3负极导电性差和循环过程中体积变化大的问题,Zheng等[73]设计和制备了碳层均匀包覆的蛋黄@壳柱状结构Fe2O3@C 复合材料。蛋黄/壳结构由于在核壳之间具有空腔被认为是缓解高比容量负极材料的体积效应进而实现优异的电化学性能的合理设计。然而,目前大多数工作是采用常规电化学测试和充放电后拆解电池的事后分析来证明蛋黄/壳结构对电极材料循环稳定性的优势,而缺乏动态监测电化学过程中活性物质在空腔的演变和相互作用及与电池循环性能间关联的深入研究。Zheng等[48]利用原位TEM实时监控和解析具有蛋黄@壳柱状结构Fe2O3@C 负极材料的微观演变和稳定机制(图10)。研究发现,在嵌锂过程中,柱状Fe2O3发生显著体积膨胀并填充Fe2O3和C 层之间的空腔[图10(a)~(c)]。由于空腔的存在,Fe2O3体积膨胀的应力得到有效释放使得外层碳壳保持完整,显著改善了电化学性能。首次完全嵌锂后,单晶Fe2O3纳米颗粒转变成了大量超细的Fe 纳米晶并镶嵌在Li2O 的基体中,跟Su 等原位TEM 观察到的结果一致。脱锂完全后,反应产物为FeO,而不是初始的Fe2O3[图10(d)~(g)],但是微观形貌跟嵌锂后没有明显变化[图10(h)]。原位TEM多次循环测试证实,空腔结构能够更有效地缓解嵌/脱锂过程中Fe2O3剧烈的体积变化,其整体结构在多次循环过程后可完好保持[图10(i)~(l)],展现出了优异的循环性能。该研究从微观层面上揭示了空腔结构抑制体积效应的机制,可为材料储锂性能、体积效应与材料多孔结构之间关系的研究提供新的实验证据和思路。

图10 (a)~(c)嵌锂过程中蛋黄壳结构Fe2O3@C电极的TEM图;第一次锂化(d)和脱锂(h)后的TEM图;第一次锂化前(e)、第一次锂化后(f)和第一次脱锂后(g)的SAED图;第二次嵌锂后(i)、第二次脱锂后(j),第三次嵌锂后(k)、第三次脱锂后(l)的TEM图[73]Fig.10 (a)~(c)Time-lapse TEM images of a yolk@shell Fe2O3@C electrode during first lithiation process;TEM images acquired after first lithiation(d),and first delithiation(h);SAED patterns before lithiation(e),after first lithiation(f)and after first delithiation(g);TEM images after second lithiation(i),after second delithiation(j),after third lithiation(k)and after third delithiation(l)[73]
过渡金属磷化物(例如磷化铁、FeP)作为一类转化型储锂机理的负极,表现出较高的理论比容量、稳定的放电平台和相对较低的极化电位,被认为是有潜力的下一代锂离子电池负极材料。然而在循环过程中也存在体积变化大、颗粒团聚、导电性差以及放电产物Li3P可逆性差等问题,易导致其容量快速衰减和倍率性能欠佳。将其镶嵌或包埋在导电碳骨架中制备出合理的纳米颗粒/碳核壳复合结构,可显著改善电极材料的循环和倍率性能。然而纳米颗粒与碳骨架在电化学过程中的动态相互作用和电化学性能增强机制,依然缺乏微观层面的直接实验佐证。Zheng等[74]通过一种简单的方法将非晶FeP 纳米颗粒封装在超薄3D 交联的磷掺杂多孔碳纳米片中(FeP@CNs),形成纳米颗粒@3D 交联的磷掺杂多孔碳纳米片包埋型复合结构,从而缓释其体积膨胀、提供电子导电性并有效抑制颗粒团聚。利用原位TEM技术实时研究了纳米颗粒和3D碳骨架间相互作用,并揭示了其结构稳定的微观成因(图11)。通过动态监测嵌锂过程中纳米颗粒的体积膨胀情况,可发现,在完全嵌锂后5个纳米颗粒的体积膨胀均在120%~130%,且颗粒可在碳片内部自由膨胀而未造成碳片破损和团聚现象[图11(a)~(c)]。上述原位TEM 表征有力证明了3D 多孔碳结构可通过交联骨架的伸缩来缓冲纳米颗粒的膨胀应力,避免了因应力造成的磷化铁颗粒粉碎和团聚,使纳米颗粒得以稳定工作并有效输出高的可逆容量。通过原位TEM技术对复合材料的多次循环进行连续观测[图11(d)~(f)],可以进一步佐证上述结论:3D交联的碳纳米片对缓解非晶FeP纳米颗粒的体积膨胀和防止其纳米颗粒在循环过程中的粉化/团聚起着至关重要的作用,有利于提高其循环性能。此外,完全嵌锂和完全脱锂状态下电极的选区电子衍射[图11(g)~(i)]、非原位TEM以及同步辐射表征也同样证明了复合电极在循环过程中发生了高度可逆的转化反应。该研究揭示了包埋型纳米颗粒/碳核壳复合结构电极在电化学过程中物相和形貌的动态演变对其电化学性能的影响规律,可为高比容量负极材料的设计提供重要科学依据。

图11 (a)~(c)嵌锂过程中FeP@CNs电极的TEM图;初始FeP@CNs电极[(d)、(g)],第一次锂化[(e)、(h)]和第一次脱锂[(f)、(i)]后的原位TEM图和相应的SAED图[74]Fig.11 (a)~(c)Time-resolved TEM images of a FeP@CNs electrode during lithiation.In situ TEM images and corresponding SAED patterns of pristine FeP@CNs electrode[(d)、(g)],after first lithiation[(e)、(h)]and first delithiation[(f)、(i)][74]
此外,针对其他转化反应负极材料(Fe3O4[75-76]、CuO[77]、 Co3O4[78]、 NiO[79]、 RuO2[80]、 FeS[81]、CoS2[82]、WO3[83]等),研究工作者利用原位TEM也研究了它们在电化学过程中的微观电化学行为、结构演变和反应机理,这些研究结果为寻找并开发性能优异的电极材料、理解其失效过程和优化其电化学性能提供了直观可靠的科学依据。
3 结论与展望
综上所述,本文总结归纳了近年来人们利用原位TEM 技术研究锂离子电池正极和高比容量负极材料所取得的重要研究进展,内容涵盖电极材料在充放电过程中的化学成分、微观结构演化、相变、尺寸效应、反应动力学及其动态演化行为等信息。进一步讨论和展望了原位TEM 技术存在的问题,以及应用这项技术研究二次电池未来研究的挑战和发展方向。我们希冀通过本文中的评述,能帮助研究人员更深入地理解原位TEM 技术研究电极材料在充放电过程中微观结构、形貌、成分动态演变过程、失效的微观机理以及与材料电化学性能间构效关系等关键基础科学问题中的应用,为构筑高能量密度、长寿命、高安全的下一代锂离子电池提供重要的理论依据和实验策略。总体而言,原位TEM技术具有:①可实现在纳米或原子尺度上动态监测电极材料在充放电过程中的微观结构、形貌和物相的动态演化过程的独特优势;②加装能量色散X射线谱(EDS)、电子能量损失谱(EELS)、扫描隧道显微镜(STM)、原子力显微镜(AFM)等,进一步实时动态分析器件或材料的元素组成、价态变化、电子输运和力学响应等信息;③利用可操纵的原位样品杆(力、热、光、电样品杆)和氮化硅窗口保护的液态样品池以及环境腔体,实现多场耦合和不同环境下的原位表征。这些优势将为更深入理解锂离子电池电极材料在真实电化学循环中的演变过程、失效机理和结构变化提供直接的科学依据。
原位TEM 在电极材料研究中具有独特优势,但仍然在动态研究、定量分析和检测分辨率等方面面临困难和挑战,需要进一步优化和创新。未来原位TEM 技术研究二次电池关键电极材料相关基础问题的发展应注重如下几个方面,包括但不限于以下几点。
(1)目前大多数原位TEM 表征在构建纳米电池时,使用锂表面自然氧化形成的氧化锂作为电解质,这与实际宏观锂离子电池中使用的电解液或电解质存在差异,因此,原位TEM 所揭示的电极现象和反应机制与实际宏观锂离子电池中的现象和机制可能有所差异。今后的研究可综合原位TEM 与非原位表征技术对电极材料进行测量,以期获得更为全面准确的信息。此外,搭建更接近电池中真实环境的原位TEM 平台肯定会有助于揭示电池性能衰减的真实机制。除了上述的全固态开放式纳米电池外,原位液体环境下的电化学原位TEM 也是发展的方向之一。
(2)几乎所有原位TEM 的纳米电池装置都只能测试少数几次充放电循环,并不能很好地揭示长循环过程中材料和界面的演变和失效机制。因此,需要开发新的原位TEM 技术,并结合加速试验,以实现长周期下的实时观测研究。
(3)电化学反应需要外加电压才能进行。然而原位TEM 测试时所施加的实际电压显著依赖于电极材料与电解质间的接触条件。因此,很难定量地确定电压及其与反应动力学的关联。为解决这一问题,可开发三电极结构的原位纳米电池装置,实现纳米级电化学的定量测量。
(4)在原位TEM 实验过程中,电子束对电化学反应的影响是不可避免的。这可能导致充放电过程中观察到的现象和机制出现干扰因素。因此,在TEM 空间有限的情况下,需要设计一个既能保证电子束传输,又能接近真实电池工作状态的反应环境。
(5)电化学循环过程中SEI 和CEI(正极-固态电解质界面)的演化是调控锂离子电池电化学性能的关键因素。然而,锂离子电池研究对SEI层的理解仍不够深入。目前,亟需开发一种适合于在电化学循环过程中,在电极与固态电解质之间原位表征SEIs 和CEIs 的形成、生长和力学性能的新型纳米级电池平台。近期冷冻电镜在电池材料中的成功应用可能会对原位研究SEI或CEI的演变有帮助。
(6)虽然原位TEM 可在纳米甚至原子尺度下表征锂离子电池电极的结构和组成信息,但大多数原位TEM 实验采用电压控制模式。由于单个纳米线或纳米微粒电极的电流非常小,在电流控制或恒电流充放电模式下研究电极材料的同步物理化学性质动态演变过程与失效机制难以实现。因此,同时动态测量锂离子电池电极材料的结构演化与宏观电化学性质也是原位TEM面临的重大挑战之一。
(7)考虑到原位TEM 研究锂离子电池电极材料微观结构演变和电化学机理的局限性,同时表征电极、电解质及其界面和中间相的时空演化仍存在较大挑战。可将多种原位技术(原位TEM、原位中子散射、原位X 射线衍射、原位拉曼等技术)/非原位技术结合,并辅以计算建模,实现对锂离子电池电极材料及界面问题更全面和深入的理解,从而获得全面准确的构效关系,进而指导新的材料与器件的设计。
猜你喜欢
汽车工程师(2021年12期)2022-01-18
汽车工程师(2021年12期)2022-01-18
陶瓷学报(2021年5期)2021-11-22
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
云南化工(2020年11期)2021-01-14
科学(2020年1期)2020-08-24
天然产物研究与开发(2018年5期)2018-06-13
中国有色金属学报(2018年2期)2018-03-26
电源技术(2016年9期)2016-02-27
火炸药学报(2014年1期)2014-03-20
