
53BP1缺失补偿DNA同源重组修复增强BCCIP阴性乳腺癌细胞放疗抗性
2021-07-07 10:16罗婉蓉金晓旎欧阳钰沭刘宁昂
激光生物学报 2021年3期
罗婉蓉,余 佳,刘 冲,金晓旎,欧阳钰沭,童 星,刘宁昂*
(1.苏州大学医学部放射医学与防护学院,放射医学与辐射防护国家重点实验室,江苏省高校放射医学协同创新中心,苏州 215123;2.苏州大学医学部实验中心,苏州 215123)
20世纪70年代末以来,全球乳腺癌发病率一直呈上升趋势。2018年,预计全国约有36.8万新发乳腺癌病例,乳腺癌已成为45岁以下女性最常见的癌症死因[1]。三阴性乳腺癌(triple negative breast cancer,TNBC)是指雌激素受体(estrogen receptor,ER)、孕激素受体(progesterone receptor,PR)及人表皮生长因子受体2(human epidermal growth factor receptor 2,HER2)三种受体均呈阴性表达的乳腺癌亚型。中国女性TNBC患者占所有乳腺癌的12%~20%,远高于西方国家10%~15%的占比[2-3]。与激素受体(ER、PR)阳性或HER2阳性乳腺癌相比,由于分子特征的限制,TNBC缺少相关的内分泌和靶向治疗靶点,导致其高复发率和高死亡率,且放疗后TNBC的局部复发率远高于其他乳腺癌亚型[4-5]。这提示TNBC存在固有的和/或获得性的放疗抗性,可能是导致TNBC放疗失败的重要原因。已有研究表明,约50%的TNBC与乳腺癌易感基因1(breast cancer susceptibility gene 1,BRCA1)及抑癌基因p53的突变或缺失相关[6-8]。然而,对于另外50%与BRCA1及p53缺陷不相关的TNBC亚型,其辐射抗性比BRCA1/p53缺陷型TNBC更高,且发病的分子机制尚不清楚[9-10]。因此,探明这一部分非BRCA1相关的TNBC辐射抗性的分子机制,是降低TNBC的辐射抗性和改善放疗预后的关键。
辐射诱导的DNA双链断裂(DNA double-strand break,DSB)是细胞死亡的重要原因。因此,细胞内DSB的修复能力是决定其辐射敏感性、基因组完整性和细胞存亡命运的关键。抑制DSB的精准修复能增加肿瘤细胞对辐射的敏感性,而细胞内过高的DSB修复活性则导致肿瘤细胞辐射抗性的增强。真核细胞主要通过两种修复方式应对DSB损伤:其一是修复精确性较低的非同源重组末端连接(nonhomologous end joining,NHEJ);其二是修复精确度极高的同源重组修复(homologous recombination,HR)。两种DSB修复途径的选择被细胞严格调控。以DSB修复相关的分子为靶标,通过对DSB修复通路的干预,选择性抑制高修复精确度的同源重组修复活性,增加DNA辐射损伤修复错误率,有可能降低细胞的辐射抗性,从而改善放疗的疗效及预后。
本研究的前期研究中,特异性敲除小鼠乳腺上皮细胞中的DNA同源重组修复相关蛋白(BRCA2 and CDKN1A interacting protein,BCCIP)后,约50%的雌鼠快速形成了乳腺结节,其中10%最终发展成乳腺癌,并且所有BCCIP缺陷的乳腺癌细胞都丧失了DSB修复关键蛋白53BP1[11]。在乳腺癌病人的病理组织样本中,约49%的TNBC中出现了BCCIP的自发缺失,同时53BP1的缺失和BCCIP阴性型TNBC呈高度相关[11]。根据上述研究的结果,本研究提出,53BP1的缺失提高了BCCIP阴性肿瘤细胞的辐射抗性。因此,基于前期大量的临床、动物试验数据及文献调研证据,本研究通过阐明BCCIP/53BP1途径对乳腺癌细胞辐射敏感性的调控机制,明确了约50%高辐射抗性TNBC的分子特征,为优化这类乳腺癌的治疗方案、发现新的放疗增敏靶点提供理论和试验依据。
1 材料与方法
1.1 主要试剂和材料
小鼠乳腺癌4T1细胞株购自中国科学院上海生命科学研究院细胞库;小鼠胚胎成纤维细胞(mouse embryonic fibroblasts,MEF)为本实验室构建;DR-GFP U2OS(人骨肉瘤细胞)由深圳大学许兴智教授提供;人TNBC细胞MDA-MB-231由苏州大学畅磊教授提供;RPMI-1640(Roswell Park Memorial Institute-1640)培养基、DMEM(Dulbecco’s modified eagle medium)培养基和胎牛血清(fetal bovine serum,FBS)购自美国Gibico公司;胰酶、青霉素/链霉素双抗和左旋谷氨酰胺购自江苏海门碧云天生物技术有限公司;着丝粒和端粒荧光探针购自韩国Panagene公司;γH2AX抗体购自美国Cell Signaling Technology公司;53BP1和Lamin B抗体购自美国Santa Cruz公司;GAPDH抗体购自苏州睿瀛生物技术有限公司;BCCIP抗体、Lipofectamine 3000转染试剂及siRNA(si53BP1)购自美国Thermofisher公司。
1.2 细胞培养及BCCIP稳定敲低细胞株的构建
4T1 细胞培养于含有10% FBS、1% 双抗及1%左旋谷氨酰胺的RPMI-1640培养基中,MEF、DRGFP U2OS和MDA-MB-231细胞均培养在含有上述相同添加成分的DMEM培养基中。细胞在37℃、5% CO2及饱和湿度的培养箱中常规培养,用0.25%的胰酶消化传代。试验所用细胞处于对数生长期。使用慢病毒载体plKD-CMV-Puro-U6-shRNA,插入文献已报道的小鼠BCCIP shRNA回文序列(5'-GGATGAAGATGAGATCTTTGGTTCAAGAGACCAAAGATCTCATCTTCATCCTTTTTT-3')[12]。配合psPAX2和pMD2.G辅助质粒转染人肾上皮细胞293T细胞包装病毒,并利用成熟的病毒感染细胞3次,以过表达shBCCIP序列构建BCCIP稳定敲低的细胞株。
1.3 细胞转染
当细胞生长至60%~80%汇合度时,使用Lipofectamine 3000转染试剂对DR-GFP U2OS细胞进行si53BP1的转染。每105个细胞使用20 pmol siRNA。4T1、MEF和MDA-MB-231细胞用胰酶处理后混入si53BP1,使用Lonza Amaxa Nucleofector II电穿孔仪protocol T-24预先设定的电压和脉冲频率进行转染。上述细胞转染48 h后收获,用于蛋白质印运(Western blot)检测或后续试验。本研究的试验分组如下表1。

表1 本研究试验分组Tab.1 The experimental groups in this study
1.4 Western blot检测
除去细胞培养皿中的培养基,用磷酸盐缓冲溶液(phosphate buffer saline,PBS)洗细胞2遍,彻底去除PBS后,加入适量十二烷基硫酸钠(sodium dodecyl sulfate,SDS)细胞裂解液,冰上裂解后收集蛋白。蛋白液经超声破碎后,100℃变性5 min,涡旋混匀后上样。分别使用12%和5%~6%的十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)对γH2AX和53BP1进行检测,电泳完成后进行转膜。转印有蛋白的聚偏二氟乙烯(polyvinylidene fluoride,PVDF)膜,经5%脱脂牛奶室温封闭2.0 h。随后,将封闭好的PVDF膜与γH2AX、BCCIP、53BP1或GAPDH等一抗在4℃条件下分别过夜孵育。第二天用PBST洗膜3次,每次10 min。室温孵育相应种属的二抗1.5 h,PBST洗膜后,用ECL(electrochemiluminescence)化学发光试剂在显影机(Proteinsimple,FluorChem M)上检测目的蛋白质条带信号。
1.5 免疫荧光染色
细胞于照射后的不同时间经4.00%多聚甲醛(paraformaldehyde,PFA)/PBS室温固定10 min,用0.50% SDS和0.25% Triton X-100/PBS室温通透5 min。随后,经1.00%小牛血清(bull serum albumin,BSA)/PBS室温封闭1 h,将样品置于湿盒内,在4℃条件下过夜孵育一抗。次日清洗PBS后用对应的荧光二抗室温孵育1.0 h。最后,经4',6-二脒基-2-苯基吲哚(4',6-diamidino-2-phenylindole,DAPI)染色并封片,在荧光显微镜下观察结果。每组独立试验至少分析200个细胞的焦点数,试验重复3次。
1.6 辐射类型及剂量
根据不同试验的需要,利用X射线仪(RS 2000X-ray Biological Irradiator,Rad Source Technologies)对细胞进行0、2、4、6 Gy剂量的 X 射线照射,剂量率为1.046 Gy/min。在细胞受照后不同时间点收集样品进行后续分析。
1.7 克隆形成试验
首先,利用野生型细胞测试不同剂量下的克隆形成率。将充分分散的细胞按照推算的克隆形成率接种于60 mm培养皿中,12 h后进行照射处理,每处理组设置3个平行样品以增加试验可靠性。照射14 d后,显微镜下观察每个克隆集落中的细胞数,当细胞数多于50个时,用甲醇固定细胞,并用结晶紫对细胞克隆进行染色,回收结晶紫溶液并清洗培养皿。待干燥后,计算细胞存活分数(surviving fraction,SF),绘制存活曲线,计算半数致死剂量(median lethal dose,LD50)。
1.8 端粒-着丝粒荧光原位杂交(CT-FISH)和姐妹染色单体(SCE)互换试验
端粒-着丝粒荧光原位杂交(telomere-centromere fluorescencein situhybridization,CT-FISH)试验:细胞经过2 Gy照射之后,立即加入0.025 μg/mL秋水仙碱,并在37℃培养 24 h以截获辐射损伤修复后的中期染色体。胰蛋白酶消化并收集细胞,使用0.075 mol/L的氯化钾低渗处理细胞使染色体松弛,随后采用经典的Carnoy固定液[V(CH3OH)∶V(CH3COOH)=3∶1]4℃固定细胞3次。在染色体铺片完成后,染色体与FITC-telomere PNA(peptide nucleic acid)探针和Cy3-centromere PNA探针(Panagene,South Korea)的混合杂交液在80℃高温下变性3 min,并室温杂交1 h,标记染色体的着丝粒和端粒,最后用DAPI标记染色体,完成CT-FISH的制片。
姐妹染色单体互换(sister chromatid exchange,SCE)试验:向细胞中加入BrdU(终浓度为10 µmol/L),并于24 h后给与细胞2 Gy照射,照射后立即加入0.025 μg/mL秋水仙碱,37℃继续培养24 h以截获辐射损伤修复后的中期染色体。胰蛋白酶消化并收集细胞后,使用0.075 mol/L的氯化钾低渗处理细胞使染色体松弛。随后,采用经典的 Carnoy 固定液[V(CH3OH)∶V(CH3COOH)=3∶1]4℃固定细胞 3次。在染色体铺片完成后,加入Hoechst 33258并在60℃条件下经black light曝光处理1 h。最后,用DAPI标记染色体,完成SCE制片。
上述制作好的玻片使用中期染色体自动捕获细胞遗传学工作站(MetaSystems/Zeiss Axio Imager Z2)进行PNA 探针标记的染色体和SCE染色体的高通量自动识别和图像拍摄。使用Isis(MetaSystems)软件对染色体进行快速分析。
1.9 数据处理与统计
每组试验至少重复3次,数据以平均值±标准差表示。使用t检验进行统计学分析,P<0.05为差异具有统计学意义。
2 结果与分析
2.1 高通量染色体快速精确分析系统的建立
基于中期染色体自动捕获细胞遗传学工作站(MetaSystems/Zeiss Axio Imager Z2)的软件和硬件平台(图1a),首先建立了采用 CT-FISH 技术的染色体快速精确分析系统。相对于传统的吉姆萨(Giemsa)染色,CT-FISH分析可以更加清楚直观地展现染色体的端粒和着丝粒(图1b),为精确分析不同类型的染色体畸变(图1c,双着丝粒染色体、三/四着丝粒染色体、片段、着丝粒环等),评价电离辐射后DSB的修复效率、基因组稳定性维持的能力等提供了优良的技术平台。
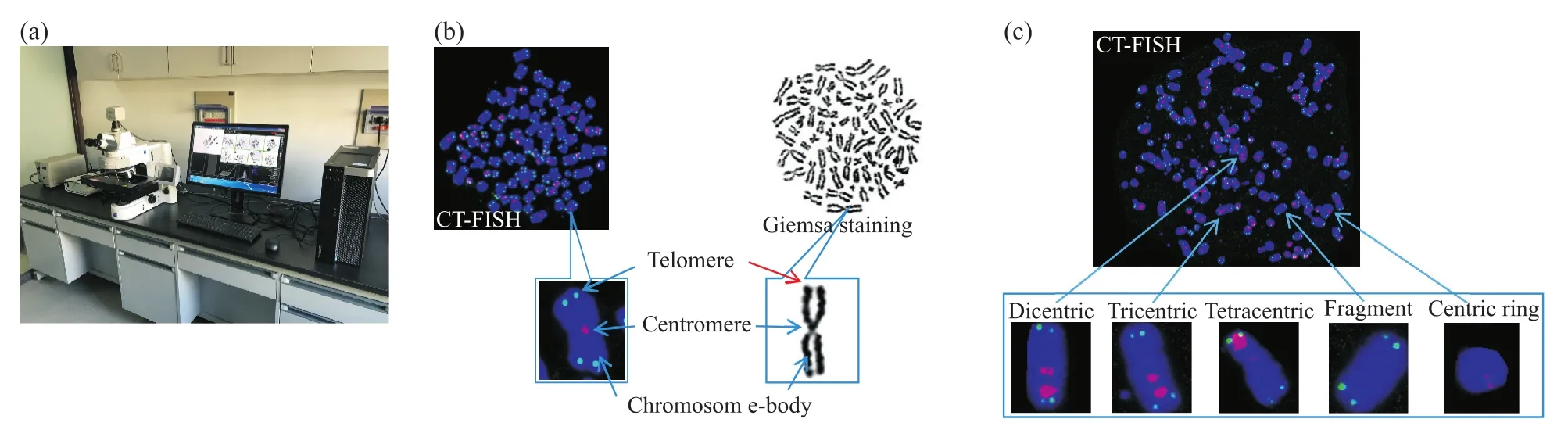
图1 中期染色体自动捕获MetaSystems/Zeiss Axio Imager Z2细胞遗传学工作站及CT-FISH对比吉姆萨染色在染色体分析中的优势Fig.1 The advantages of metaphase chromosome automatical capture cytogenetics workstation established by MetaSystems/Zeiss Axio Imager Z2 and CT-FISH technique in chromosome aberration analysis compared to Giemsa staining approach
2.2 53BP1缺失增加BCCIP阴性乳腺癌细胞辐射后基因组稳定性
为明确53BP1的同时缺失是否提高了BCCIP阴性乳腺癌细胞对辐射基因毒性损伤的抵抗能力,本研究在验证了53BP1和BCCIP的敲低效率后(图2a),首先应用高通量染色体快速精确分析系统检测53BP1缺失对BCCIP阴性4T1乳腺癌细胞电离辐射导致异常染色体形成率的影响,鉴定53BP1对辐射损伤后BCCIP阴性细胞基因组稳定性维持能力的调节作用。分析结果显示,BCCIP的单独缺失导致辐射后三着丝粒染色体畸变率的显著增加(图2b),这一结果与之前报道的BCCIP参与同源重组修复相符合[12]。而53BP1在BCCIP阴性4T1乳腺癌细胞中的额外缺失可以下调BCCIP缺陷导致的辐射后染色体高畸变率(图2b),提示53BP1的额外缺失增加了辐射后细胞基因组稳定性的维持功能,部分恢复了BCCIP阴性细胞对辐射遗传毒性的抵抗能力。
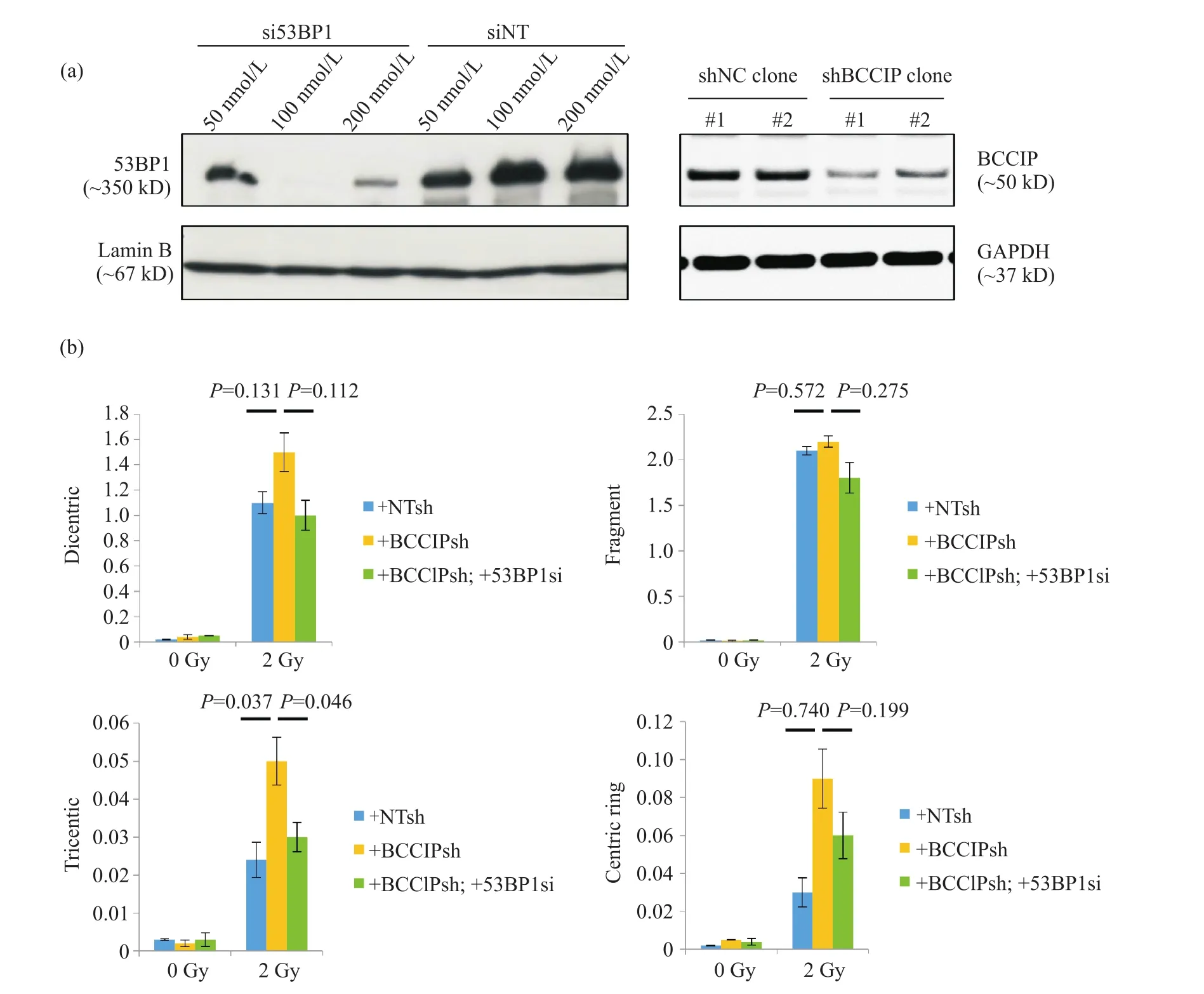
图2 53BP1缺失可部分恢复BCCIP阴性细胞对辐射遗传毒性的抵抗Fig.2 The defected resistant capacity to radiation genotoxicity in BCCIP deficient cells can be partially restored by 53BP1 additionally deletion
2.3 53BP1敲低促进BCCIP阴性乳腺癌细胞中辐射诱导DNA双链断裂的修复能效
辐射可以引起细胞基因组DNA多种类型的损伤,其中DSB是最为严重且致命的DNA损伤类型。第139位丝氨酸磷酸化修饰后的组蛋白H2AX被称之为γH2AX,是公认的DNA双链断裂标志物,而辐射后γH2AX焦点的聚集形成被认为是细胞识别DSB并做出反应的关键步骤。根据细胞种类的不同,γH2AX水平一般在细胞受照0.5~2.0 h之间达到峰值,随后由于细胞DNA修复程序的启动而逐渐降低,约在受照8.0~24.0 h之间基本完成主要的DSB修复工作。因此,应用2 Gy X射线照射4T1细胞和MEF细胞诱导DSB损伤,并在照射后4.0 h固定细胞,通过免疫荧光染色分析γH2AX 焦点数量可以作为细胞DSB修复效率的评价依据。结果显示,未照射的BCCIP和53BP1阳性的野生型细胞几乎没有γH2AX焦点形成,而BCCIP单缺失细胞则存在自发DSB损伤水平的升高(无统计学差异)。另外,与野生型细胞相比较,BCCIP阴性细胞经2 Gy剂量照射后,γH2AX 焦点数在照射后4.0 h依然保持较高水平,而53BP1的额外缺失可以明显降低BCCIP阴性细胞中的γH2AX 焦点数量(图3a)。这些结果提示,53BP1的同时缺失可以促进BCCIP阴性乳腺癌细胞受照后的DSB损伤修复能力。另外,我们在 BCCIP 敲低的MEF细胞中抑制53BP1表达,利用Western blot发现,细胞在4 Gy X射线照射8.0 h后γH2AX水平较BCCIP 单缺失组变弱,更接近正常对照组损伤修复后的恢复水平,提示53BP1缺失可以改善辐射后BCCIP阴性细胞中DSB的修复效率(图3b)。与以往文献报道相符,BCCIP单缺失组因同源重组修复效率的明显抑制,自发及诱发DSB均无法得到有效修复,因而使细胞维持较高的γH2AX活化状态(图3)。

图3 53BP1缺失促进BCCIP阴性乳腺癌细胞DSB修复效率Fig.3 The DSB repair efficiency in BCCIP negatively expressed breast cancer cells was enhanced by 53BP1 concurrent deletion
2.4 53BP1调控BCCIP低表达细胞同源重组修复及细胞辐射抗性
DR-GFP系统是利用细胞的同源重组修复,以特定序列为模板,将被I-SceI内切酶破坏的不完整GFP序列修复成具有表达活性的完整GFP荧光蛋白,从而通过荧光信息的表达情况报告细胞同源重组修复的效率。我们在DR-GFP U2OS细胞中同时敲低 BCCIP 与 53BP1,并转入I-SceI内切酶表达质粒pCBAS,在GFP序列产生特异性的DSB位点,随后利用流式细胞仪分析经同源重组修复后GFP荧光阳性细胞的比例。如图4a所示,与BCCIP/53BP1阳性的DR-GFP U2OS细胞相比,BCCIP单敲低导致GFP信号的明显降低,而53BP1在BCCIP敲低细胞中的同时缺失则显著恢复了GFP荧光信号,提示53BP1的缺失可以恢复BCCIP阴性细胞的同源重组修复效率。
由于同源重组修复是介导DSB损伤后姐妹染色单体互换的重要途径,因此姐妹染色单体的互换率也被广泛作为评价同源重组修复效率的指标。利用Metasystem高通量染色体快速精确分析系统检测姐妹染色单体互换率,得出与DR-GFP试验相似的结果,53BP1缺失可以显著补偿辐射损伤后BCCIP敲低造成的同源重组修复效率的抑制(图4b)。
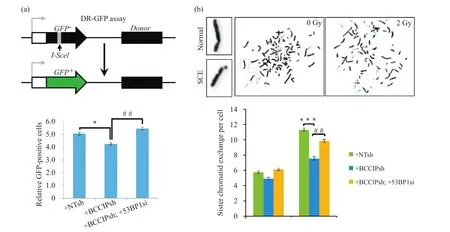
图4 53BP1缺失恢复BCCIP阴性细胞同源重组修复效率Fig.4 53BP1 depletion restored homologous recombination repair efficiency in BCCIP deficient cells
2.5 53BP1缺失增强BCCIP低表达乳腺癌细胞的辐射抗性
克隆形成是放射生物学中最为经典的细胞辐射敏感性检测和评价指标之一。通过克隆形成试验在BRCA1正常型人TNBC细胞MDA-MB-231中发现,不同剂量照射后,同时敲低BCCIP和53BP1的细胞克隆形成率较 BCCIP 单缺失的细胞恢复明显(图5),提示 53BP1缺失增强了BCCIP阴性乳腺癌细胞的辐射抗性。
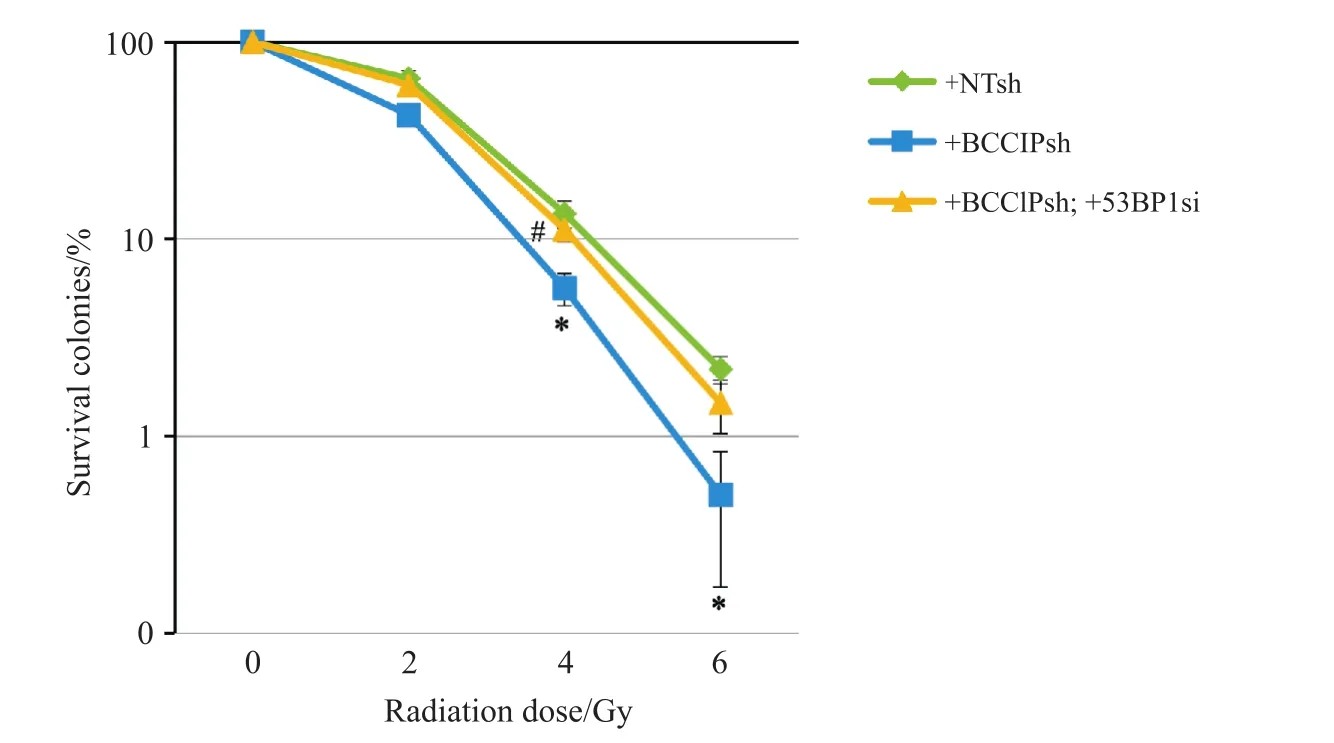
图5 克隆形成试验Fig.5 Colony formation assay
3 讨论
同源重组修复相关蛋白BCCIP于2000—2001年由两个独立的实验室发现,因其与同源重组修复关键蛋白乳腺癌易感基因2(breast cancer susceptibility gene 2,BRCA2)相互作用而得名[13-14]。研究表明,BCCIP通过影响同源重组修复效率,在肿瘤细胞辐射敏感性的调节及基因组稳定性的维持中发挥重要作用。BCCIP在神经胶质瘤、喉癌、卵巢癌、结直肠癌、肾癌、肝癌等肿瘤组织中的表达量均下降或不表达[14-19],提示BCCIP的缺失与多种实体瘤的发生相关。例如:在喉癌组织中p53表达正常,而 BCCIP缺失的病人具有较高的辐射抗性、局部复发率以及较低的生存率[16]。我们前期通过临床研究也发现,在BRCA1及p53正常的高辐射抗性TNBC中,BCCIP的表达量显著降低[11]。在473例乳腺癌病人活检样本中,有33%的样本组织检测出BCCIP表达的下调或缺失。在TNBC中,BCCIP的缺失率高达约49%,而非三阴性乳腺癌中BCCIP的缺失率仅为25%(P=3.86×10–7)。这些研究结果提示,BCCIP的表达缺失是BRCA1/p53正常型TNBC的一个重要分子特征,有可能是导致TNBC高辐射抗性的主要分子事件。
在细胞水平,BCCIP的缺失主要通过影响同源重组修复中的关键蛋白RAD51和BRCA2的功能,降低同源重组修复的效率而导致基因组不稳定,从而增加癌变风险[20-23]。与此同时,BCCIP缺失还会引起细胞对辐射的敏感性增加[24]。一个有趣的现象是,在正常细胞中,下调BCCIP蛋白会导致辐射敏感性增加,但BCCIP缺陷的肿瘤细胞却表现出高辐射抗性。为了解释这一现象,我们在前期研究中发现,约50%的组织特异性BCCIP敲低雌鼠在1年内快速形成了良性的乳腺结节,其中10%最终发展成了恶性乳腺癌[11]。更重要的是,所有BCCIP缺陷的恶性乳腺癌细胞都丧失了DNA损伤修复的一个关键蛋白——53BP1。与此同时,我们还发现,在TNBC病人的组织样本中,53BP1的缺失和BCCIP阴性肿瘤呈高度相关。根据这些临床病例研究和小鼠试验的结果,我们推测53BP1的缺失很可能提高了BCCIP阴性肿瘤细胞的辐射抗性。
接下来的问题是,53BP1缺失通过何种途径才能提高BCCIP阴性肿瘤细胞的辐射抗性?已知对DSB断裂位点3'末端局部切除修饰启动的调控在DSB修复通路的选择中起决定性作用。同源重组修复需要核酸酶在断裂末端切除修饰形成一个200 bp左右的单链同源臂。细胞周期严格调控的某些同源重组修复早期启动因子,如BRCA1、BLM(bloom syndrome protein)、Exo1(exonuclease 1)等,在DSB末端切除过程中发挥重要作用。在非同源重组末端连接中这类末端切除修饰则被抑制,DSB修复关键调控蛋白53BP1在DSB位点的迅速聚集保护其末端免于过度切除,从而决定细胞DSB损伤的修复方式,并参与调节不同DSB修复途径在细胞周期各时相中的优势选择[25-28]。病理情况下,同源重组修复相关蛋白的失活将导致细胞自发基因突变率的升高、基因组稳定性的降低,从而促进肿瘤发生率的升高。近年来,聚ADP核糖聚合酶(poly ADP-ribose polymerase,PARP)抑制剂由于对多种同源重组修复缺陷型肿瘤具有较好的治疗效果,因而被美国食品及药物管理局(food and drug administration,FDA)批准,并已广泛应用于BRCA1缺陷型乳腺癌。然而已有临床和基础研究报道提示,部分BRCA1缺陷型三阴性乳腺癌对PARP抑制剂具有较高的耐药性,其机理可能与53BP1的协同缺失及其引起的同源重组修复能效恢复有关[29-30]。如图6所示,当53BP1和BRCA1(或BLM、Exo1等)功能均正常时,细胞中的BRCA1会识别DSB位点并抑制53BP1与其结合,促进DSB末端切除复合体(DNA end resection complex,ERC)等功能蛋白对受损DNA末端的识别和切除,从而启动同源重组修复。如果同源重组早期启动因子缺失,而53BP1正常表达,由于53BP1对DSB末端切除功能的抑制,同源重组修复缺少必要的启动条件,DSB将主要通过非同源重组末端连接途径进行易错修复,导致辐射敏感性和基因组不稳定性的增加,细胞死亡。如果同源重组修复早期启动因子和53BP1同时缺失,由于53BP1抑制作用解除,同源重组修复功能得到补偿,细胞对辐射的抗性也随之增强[28-30]。巧合的是,我们在前期工作中发现,53BP1的缺失还与BCCIP阴性的TNBC高度相关[11]。因此我们提出假设,53BP1缺失通过补偿BCCIP缺陷造成的同源重组修复效能降低来维持基因组的稳定性,从而提高细胞的辐射抗性。

图6 BRCA1和53BP1共同缺失导致部分恢复辐射引起的同源重组修复[7]Fig.6 Co-deficiency of BRCA1 and 53BP1 resulted in restoration of homologous recombination repair[7]
为验证这一假说,本研究在小鼠乳腺癌等多种细胞中首先构建稳定敲低BCCIP的细胞株,在此基础上通过siRNA敲低53BP1,构建BCCIP/53BP1双缺失细胞。通过PNA荧光探针FISH技术联合高通量自动捕获系统对辐射诱导染色体畸变进行大规模分析,结果表明,敲低BCCIP引起细胞辐射后染色体畸变率的显著增加,这一结果与先前的文献报道相符[12]。而53BP1的额外缺失则降低了BCCIP阴性乳腺癌细胞染色体高畸变率的发生,从而增加辐射后细胞基因组稳定性的维持能力。随后,通过对辐射后DSB损伤标志物γH2AX的检测发现,53BP1的缺失促进了BCCIP阴性乳腺癌细胞DSB修复能力。进一步利用两种不同的DSB修复检测方法均发现,53BP1缺失通过补偿BCCIP阴性细胞的同源重组修复效率,进而提高细胞应对DSB损伤的能力。最后,通过克隆形成试验证实了53BP1的缺失增强了BCCIP阴性细胞的辐射抗性。这些结果提示,BCCIP/53BP1双缺失型TNBC的高辐射抗性可能通过丢失53BP1使BCCIP阴性细胞解除对同源重组修复的抑制,进而增加BCCIP阴性乳腺癌细胞同源重组修复的效率,最终增强了细胞的放疗抗性。
综上所述,本研究从BCCIP/53BP1这条新途径阐明TNBC高辐射抗性的原因,明确了BCCIP/53BP1双缺失乳腺癌细胞高放疗抗性是通过53BP1的缺失增强了BCCIP阴性细胞DSB损伤的同源重组修复能力。目前已报道的BRCA1/p53途径只能解释约50% TNBC辐射敏感性的调节机制,而本研究的发现说明了另外50% TNBC辐射抗性更强的原因,从而为全面阐明TNBC高辐射抗性的分子机制,寻找新的潜在放疗增敏分子靶标,提供了理论基础和试验依据。
猜你喜欢
华人时刊(2023年1期)2023-03-14
汉字汉语研究(2021年2期)2021-08-30
空间科学学报(2021年1期)2021-05-22
汉字汉语研究(2019年2期)2019-08-27
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
环境保护与循环经济(2017年5期)2018-01-22
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
河北书画研究(2016年3期)2016-04-28
中国果菜(2016年9期)2016-03-01
