
玫瑰红景天活性成分络塞维的合成研究
2021-07-06 14:05钟婷胡亚丹苏进杨淑珍何贝桥张园园
中国现代中药 2021年5期
钟婷,胡亚丹,苏进,杨淑珍,何贝桥,张园园
北京中医药大学 中药学院,北京 102488
络塞维为肉桂醇基-6-O-(α-L-吡喃阿拉伯糖基)-β-D-吡喃葡萄糖苷(图1),是由肉桂醇与蚕豆糖所构成的二糖苷。络塞维是天然抗氧化中药玫瑰红景天RhodiolaroseaL.的活性成分和指标性成分[1]。据文献报道,其具有抗疲劳、抗辐射、抗氧化、抗癌、增强免疫、改善学习记忆能力等多种药理活性[2-5],应用前景广阔。络塞维主要是从玫瑰红景天中提取、分离、纯化后制备[6],但质量分数仅为0.21%~0.55%[1],使得分离纯化难度大、成本高,有必要进行化学合成。目前,络塞维的合成方法主要有2种[7-9]:1)Msashi等[7]将D-葡萄糖与丙烯醇通过固定化β-葡萄糖苷酶催化成苷,然后通过保护与脱保护策略,得到烯丙基-2,3,4-三-O-苯甲酰基-β-D-吡喃葡萄糖苷,再与溴代阿拉伯糖反应,最后与苯基硼酸反应,脱苯甲酰基保护基得络塞维。但该方法使用的丙烯醇为剧毒、管制药品,醋酸钯和固定化β-葡萄糖苷酶价格昂贵,且需经过2次苯甲酰基保护的二糖苷的分离,操作烦琐。2)另一种思路是通过糖6位羟基暴露的D-吡喃葡萄糖受体与L-吡喃阿拉伯糖供体合成二糖中间体,再将其制备成二糖供体与肉桂醇进行糖苷化反应,最后脱保护基得络塞维。但是在二糖中间体合成过程中副产物多,分离困难,且进一步制备二糖供体时收率大大降低,很难得到。
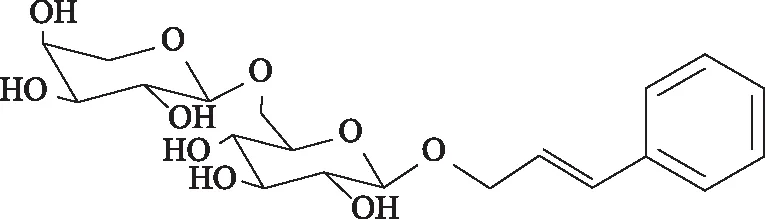
图1 络塞维结构
本研究以肉桂醇为原料,与全乙酰-α-D-溴代葡萄糖进行成苷反应,经脱乙酰基保护获得肉桂醇基-β-D-吡喃葡萄糖苷,接着用三苯基氯甲烷对糖的6′位羟基选择性保护、其他位羟基乙酰化保护及脱糖6′位保护基,特异性地将糖6′位伯羟基暴露,再与全乙酰-β-L-溴代吡喃阿拉伯糖成苷,最后脱乙酰保护基得到络塞维,路线见图2。本方法充分考虑糖类化合物合成过程中的区域选择性和立体选择性,收率较高,且未见文献报道。获得的产物和关键中间体可用于相关化合物的深入研究。本研究还可为苯丙素糖苷类化合物的制备提供参考,有助于该类天然产物在医药领域的进一步应用。
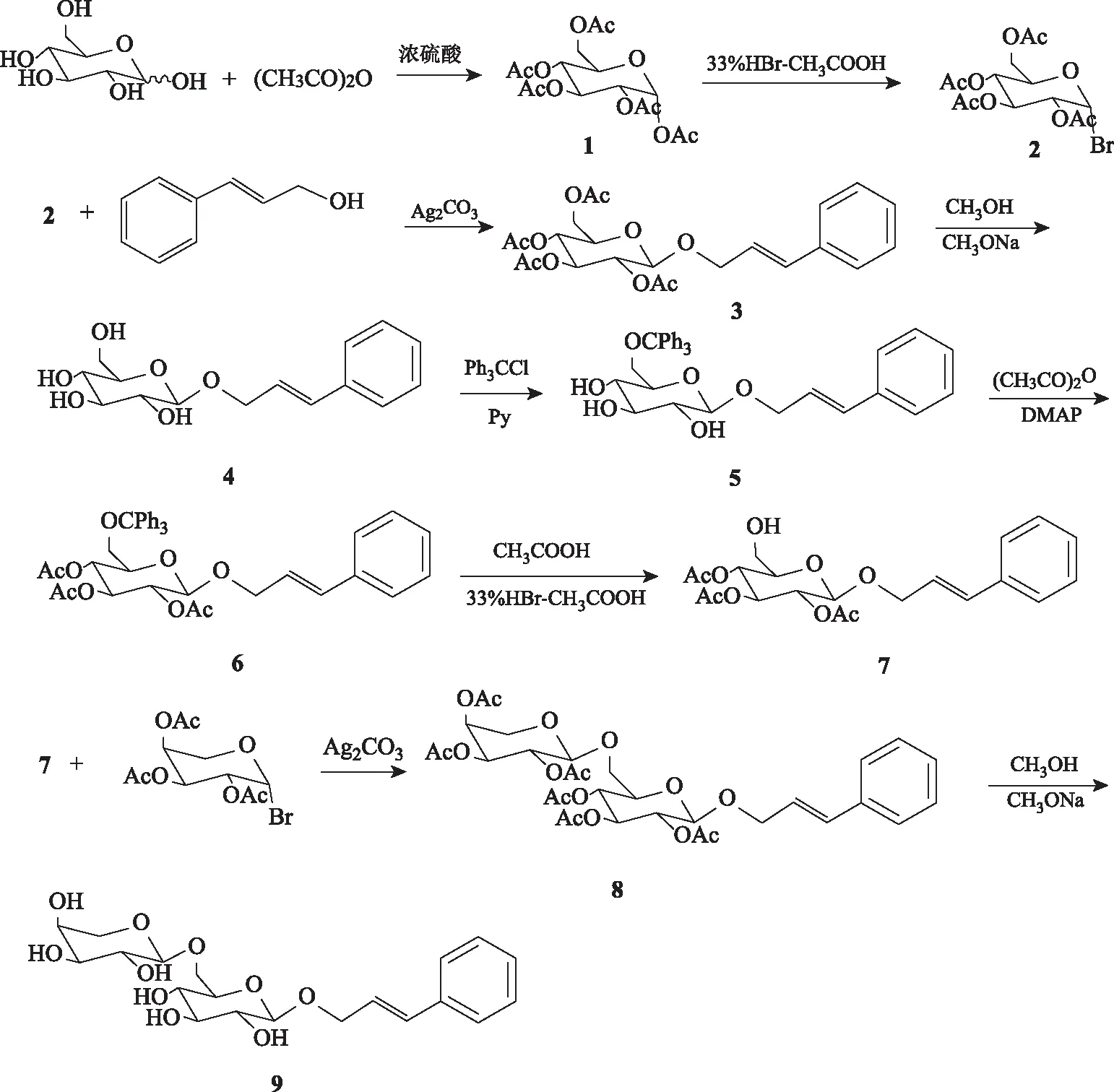
注:1.1,2,3,4,6-五-O-乙酰基-α-D-吡喃葡萄糖;2.2,3,4,6-四-O-乙酰基-α-D-溴代吡喃葡萄糖;3.肉桂醇基-2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖苷;4.肉桂醇基-O-β-D-吡喃葡萄糖苷;5.肉桂醇基-6-O-三苯基甲基-β-D-吡喃葡萄糖苷;6.肉桂醇基-2,3,4-三-O-乙酰基-6-O-三苯基甲基-β-D-吡喃葡萄糖苷;7.肉桂醇基-2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖苷;8.肉桂醇基-2,3,4-三-O-乙酰基-6-O-(2,3,4-三-O-乙酰基-α-L-吡喃阿拉伯糖基)-β-D-吡喃葡萄糖苷;9.络塞维。图2 络塞维的合成路线
1 材料
ASCEND-400型核磁共振仪(瑞士Bruker公司);WZZ-2B型半自动旋光仪(上海精密科学仪器有限公司);RE-52AA型旋转蒸发仪(上海亚荣生化仪器厂);JM-B5002型电子天平(余姚市纪铭称重校验设备有限公司);X-5型显微熔点测定仪(巩义市科瑞仪器有限公司);LC-20AT型制备液相色谱仪(日本岛津公司);Cosmosil 5C18AR-Ⅱ型制备色谱柱(250 mm×10 mm,5 μm,日本Nacalai Tesque公司)。
浓硫酸、乙酸酐(纯度:97%)、分析纯甲醇(北京化工厂);甲醇钠(纯度:98%,Acros Organics公司);33%溴化氢乙酸溶液(33% HBr-CH3COOH)、三苯基氯甲烷(纯度:98%,北京伊诺凯科技有限公司);Ag2CO3(纯度:99%,天津福晨化学试剂厂);肉桂醇(纯度:98%,阿拉丁试剂上海有限公司);D-葡萄糖、L-阿拉伯糖(纯度均为98%,阿达玛斯试剂有限公司);薄层色谱(TLC)板G(青岛海洋化工有限公司)。
2 方法与结果
2.1 1,2,3,4,6-五-O-乙酰基-α-D-吡喃葡萄糖(1)的合成
取乙酸酐11.5 mL、浓硫酸30 μL,置于圆底烧瓶中,冰浴条件下,加入D-葡萄糖4.0 g,搅拌反应1 h,撤除冰浴,室温继续反应3 h,TLC监测至反应完全。反应液用二氯甲烷50 mL萃取3次,收集、合并有机相并用水洗至中性,无水硫酸钠干燥,滤除硫酸钠后,将有机相减压浓缩得白色粉末,收率为93.7%,熔点为112~114 ℃,与文献报道一致[10]。
2.2 2,3,4,6-四-O-乙酰基-α-D-溴代吡喃葡萄糖(2)的合成
取化合物18.0 g,用二氯甲烷10 mL搅拌溶解,加入33%HBr-CH3COOH溶液5 mL,室温反应2~3 h,TLC监测至反应完全。反应液分散于适量冰水中,收集有机相,水相用二氯甲烷15 mL萃取3次,合并有机相并用水洗至中性,无水硫酸钠干燥,滤除硫酸钠后,减压回收溶剂,甲基叔丁基醚结晶,得白色晶体,收率为87.6%,熔点为86~88 ℃,与文献报道一致[11]。
2.3 肉桂醇基-2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖苷(3)的合成
取肉桂醇1.3 g,用二氯甲烷20 mL搅拌溶解,加入Ag2CO32.0 g、化合物26.6 g,避光室温反应过夜,TLC监测至反应完全,抽滤,滤饼用二氯甲烷洗1次,合并滤液,减压回收溶剂,得粗产物,直接投入2.4项下反应。
2.4 肉桂醇基-O-β-D-吡喃葡萄糖苷(4)的合成
将化合物3粗产物以5倍量甲醇于圆底烧瓶中搅拌溶解,加入适量甲醇钠调pH 8~9,室温反应4 h,TLC监测至反应完全,加适量阳离子交换树脂调pH 6~7,滤除阳离子交换树脂,减压回收溶剂,柱色谱分离(二氯甲烷-甲醇,12∶1),得白色粉末,收率为54.7%,熔点为98~99 ℃。1H-NMR(CD3OD,400 MHz)δ:7.40 (2H,d,J=7.2 Hz,H-5,9),7.28(2H,t,J=7.6 Hz,H-6,8),7.20(1H,t,J=7.2 Hz,H-7),6.67(1H,d,J=16.0 Hz,H-3),6.37(1H,dt,J=6.0,16.0 Hz,H-2),4.55(1H,dd,J=13.2,5.6 Hz,H-1),4.43(1H,d,J=7.6 Hz,H-1′),4.33(1H,dd,J=12.4,6.4 Hz,H-1),3.93(1H,dd,J=11.6,1.6 Hz,H-6′),3.78(1H,dd,J=12.0,5.2 Hz,H-6′),3.52~3.30(4H,m,H-2′~5′)。13C-NMR(CD3OD,100 MHz)δ:136.8(C-4),132.5(C-3),128.4(C-6,8),127.5(C-7),126.3(C-5,9),125.4(C-2),102.0(C-1′),76.7(C-3′),76.5(C-5′),73.7(C-2′),70.2(C-4′),69.6(C-1),61.5(C-6′)。以上数据与文献报道一致[12]。
2.5 肉桂醇基-6-O-三苯基甲基-β-D-吡喃葡萄糖苷(5)的合成
参考文献[13]方法,取化合物41.0 g置于圆底烧瓶中,用吡啶5 mL搅拌溶解,加入三苯基氯甲烷1.4 g,于50 ℃下反应7 h,TLC监测至反应完全,降温至室温后投入2.6项下反应。
2.6 肉桂醇基-2,3,4-三-O-乙酰基-6-O-三苯基甲基-β-D-吡喃葡萄糖苷(6)的合成
向2.5项下体系中加入乙酸酐1.9 mL、4-二甲氨基吡啶(DMAP)20 mg,室温搅拌过夜,TLC监测至反应完全,将反应液分散于适量冰水中,用二氯甲烷10 mL萃取3次,合并有机相,冰水和适量的稀盐酸各洗2次至弱酸性,最后用冰水洗至中性,无水硫酸钠干燥,滤除硫酸钠后,减压回收溶剂,柱色谱分离(石油醚-乙酸乙酯,5∶1),得黄色糖浆状物质,收率为76.4%。1H-NMR (DMSO-d6,400 MHz)δ:7.44 (8H,d,J=7.6 Hz,H-5,9,3″,7″,9″,13″,15″,19″),7.34(8H,dd,J=7.2,14.8 Hz,H-6,8,4″,6″,10″,12″,16″,18″),7.26 (4H,t,J=7.2 Hz,H-7,5″,11″,17″),6.68(1H,d,J=16 Hz,H-3),6.41(1H,dt,J=5.6,16.0 Hz,H-2),5.25(1H,t,J=9.6 Hz,H-3′),5.18(1H,t,J=9.6 Hz,H-4′),4.96(1H,t,J=8.0 Hz,H-2′),4.92(1H,d,J=8.0 Hz,H-1′),4.51(1H,dd,J=5.2,13.2 Hz,H-1),4.36(1H,dd,J=6.0,13.6 Hz,H-1),3.91~2.87(3H,m,H-5′,6′),2.05(3H,s,glc-4′-OAc),1.95(3H,s,glc-3′-OAc),1.72(3H,s,glc-2′-OAc)。13C-NMR(DMSO-d6,100 MHz)δ:170.1(glc-2′-OAc),169.6(glc-3′-OAc),169.1(glc-4′-OAc),143.9(C-2″,8″,14″),136.7(C-4),132.4(C-3),129.1(C-6,8),128.7(C-4″,6″,10″,12″,16″,18″),128.4~128.3(C-7,3″,7″,9″,13″,15″,19″),127.5(C-5″,11″,17″),126.9(C-5,9),126.0(C-2),99.3(C-1′),86.3(Ph3-C),73.0(C-3′),72.4(C-5′),71.7(C-2′),69.4(C-4′),68.6(C-1),61.8(C-6′),20.9(glc-4′-OAc),20.8(glc-2′-OAc),20.6(glc-3′-OAc)。
2.7 肉桂醇基-2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖苷(7)的合成
取化合物61.0 g用二氯甲烷10 mL搅拌溶解,加入冰醋酸2.5 mL,于-20 ℃下滴加33% HBr-CH3COOH溶液195.8 μL,并在此温度下继续反应2 h,TLC监测至反应完全。将反应液分散于适量冰水中,收集有机相,水相用二氯甲烷5 mL萃取3次,合并有机相,并用冰水洗至中性,无水硫酸钠干燥,滤除硫酸钠后,减压回收溶剂,柱色谱分离(石油醚-乙酸乙酯,2∶1),得无色糖浆状物质,收率为70.8%。1H-NMR (CDCl3,400 MHz)δ:7.38(2H,d,J=7.2 Hz,H-5,9),7.33(2H,t,J=7.2 Hz,H-6,8),7.26(1H,t,J=7.2 Hz,H-7),6.60(1H,d,J=16.0 Hz,H-3),6.23(1H,dt,J=6.4,16.0 Hz,H-2),5.27(1H,t,J=9.6 Hz,H-3′),5.07(1H,t,J=9.6 Hz,H-4′),4.99(1H,dd,J=8.0,9.6 Hz,H-2′),4.66(1H,d,J=8.0 Hz,H-1′),4.54~3.53(5H,m,H-1,5′,6′),2.12(3H,s,glc-2′-OAc),2.07(3H,s,glc-4′-OAc),2.06(3H,s,glc-3′-OAc)。13C-NMR(CDCl3,100 MHz)δ:171.7(glc-2′-OAc),170.3(glc-3′-OAc),169.6(glc-4′-OAc),136.4(C-4),133.0(C-3),128.7(C-6,8),128.0(C-7),126.5(C-5,9),124.7(C-2),99.7(C-1′),75.5(C-3′),74.1(C-5′),71.5(C-2′),69.9(C-4′),68.9(C-1),63.0(C-6′),20.8(glc-2′-OAc),20.8(glc-4′-OAc),20.6(glc-3′-OAc)。
2.8 肉桂醇基-2,3,4-三-O-乙酰基-6-O-(2,3,4-三-O-乙酰基-α-L-吡喃阿拉伯糖基)-β-D-吡喃葡萄糖苷(8)的合成
取化合物70.4 g,用二氯甲烷6 mL搅拌溶解,加入Ag2CO30.2 g、2,3,4-三-O-乙酰基-β-L-溴代吡喃阿拉伯糖0.5 g(制备同2.1、2.2项下方法),避光室温反应过夜,TLC监测至反应完全,抽滤,滤饼用二氯甲烷洗1次,减压蒸干溶剂,制备液相色谱法分离(甲醇-水,7∶3),得化合物8,收率为62.7%。1H-NMR(CDCl3,400 MHz)δ:7.32(2H,d,J=7.2 Hz,H-5,9),7.26(2H,t,J=7.2 Hz,H-6,8),7.18 (1H,t,J=7.2 Hz,H-7),6.55(1H,d,J=16.0 Hz,H-3),6.37(1H,dt,J=5.2,15.6 Hz,H-2),5.18~4.85(6H,m,H-2′~4′,2″~4″),4.52(1H,d,J=8.0 Hz,H-1′),4.41(1H,d,J=6.8 Hz,H-1″),4.45~4.40(1H,m,H-1),4.21(1H,dd,J=6.4,13.2 Hz,H-1),3.96~3.49(5H,m,H-5′,5″,6′),2.06(3H,s,ara-4″-OAc),1.99(3H,s,glc-4′-OAc),1.98(3H,s,ara-3″-OAc),1.97(3H,s,glc-3′-OAc),1.95(3H,s,glc-2′-OAc),1.92(3H,s,ara-2″-OAc)。13C-NMR(CDCl3,100 MHz)δ:170.2(glc-2′-OAc),170.2(glc-3′-OAc),170.1(ara-2″-OAc),169.5(ara-3″-OAc),169.4(glc-4′-OAc),169.4(ara-4″-OAc),136.4(C-4),133.0(C-3),128.6(C-6,8),127.9(C-7),126.5(C-5,9),124.4(C-2),100.8(C-1″),99.3(C-1′),73.2(C-3′),72.9(C-5′),71.4(C-2′),70.0(C-3″),69.5(C-2″),69.1(C-4′),69.0(C-1),67.9(C-4″),67.5(C-6′),63.1(C-5″),20.9(glc-2′-OAc),20.8(ara-2″-OAc),20.7(glc-4′-OAc),20.7(ara-4″-OAc),20.6(glc-3′-OAc),20.6(ara-3″-OAc)。
2.9 络塞维(9)的合成
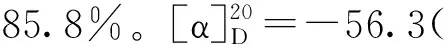
3 讨论
3.1 化合物1的合成
以葡萄糖为原料、浓硫酸为催化剂、乙酸酐为酰化试剂,合成全乙酰-α-D-吡喃葡萄糖。在D-葡萄糖投料4.0 g、浓硫酸用量为30 μL的条件下,以室温反应完全所需时间和收率为指标,考察葡萄糖-乙酸酐投料比分别为1∶3.5、1∶4.5、1∶5.5、1∶7.0。结果表明,化合物1收率分别为65.6%、91.9%、93.7%、94.6%,反应时间分别为18.0、5.0、3.0、2.5 h。综合成本和效率,以葡萄糖-乙酸酐1∶5.5为最佳条件。
3.2 化合物2的合成
用33% HBr-CH3COOH溶液对化合物1进行溴代反应,葡萄糖1.5 g用33% HBr-CH3COOH溶液1 mL反应完全,且产率较高。溴代糖常温易分解,后处理需在低温下进行,用甲基叔丁基醚结晶。
3.3 化合物4的合成
将2.3项下粗产物在碱性条件下与甲醇进行酯交换反应,不同用量的甲醇对反应完成时间和收率有影响,甲醇用量增加,反应时间缩短,产率提高。综合成本和效率,选用5倍量甲醇进行反应。
3.4 化合物8的合成
化合物7在Ag2CO3催化下与2,3,4-三-O-乙酰基-β-L-溴代吡喃阿拉伯糖进行亲核取代反应,合成糖苷,考察各反应物的投料比对产物收率的影响。结果表明,随着Ag2CO3和溴代阿拉伯糖投料量的增加,反应收率增加,但变化幅度不大。从节约成本的角度,选择化合物7-Ag2CO3-三乙酰溴代阿拉伯糖投料比为1∶0.75∶1.5,收率为62.7%。
3.5 化合物9的合成
化合物9的合成同化合物4的合成,用甲醇和甲醇钠脱乙酰基保护,但为了防止脱保护过程中二糖链断开,应加大甲醇的用量(10倍量),使其在短时间内快速反应完全。
4 结论
近年来,研究者们逐渐认识到寡糖及糖苷类化合物的生物学意义,寡糖类化合物的化学合成技术也快速发展。大量的天然产物的糖基部分存在龙胆二糖、芸香糖、蚕豆糖等1→6连接二糖,但是合成报道较少,因此,探索区域选择性和立体选择性高的路线合成1→6连接二糖具有一定的研究意义。
本研究在合成络塞维的过程中,用三苯基氯甲烷对葡萄糖的6′位伯羟基进行选择性保护,脱保护后6′位羟基裸露从而合成1→6连接糖苷键。三苯基氯甲烷基团庞大,使用其作保护基团具有选择性高、保护基团产物稳定和保护基团容易脱去的优点。本研究使用三苯基氯甲烷作为保护基团合成络塞维具有创新性,以期能够为相关二糖的合成研究工作提供新思路。
络塞维有广泛的生物学活性,本研究制备糖基供体后,在成苷反应中通过7步合成络塞维,总产率为15.92%。与文献相比,虽然未能缩短合成步骤,但实验成本降低,且条件温和、操作简单,对人体的危害和环境的污染影响较小,具有良好的工业化前景。需要说明的是,在化合物8的合成过程中,由于样品量较少,采用了制备液相进行分离,如后期大量制备可进一步探索硅胶柱色谱法进行分离。
猜你喜欢
世界农药(2022年10期)2022-11-10
林产化学与工业(2022年3期)2022-07-06
云南化工(2022年2期)2022-03-18
家庭医药·快乐养生(2022年2期)2022-02-19
能源化工(2021年2期)2021-12-30
农药科学与管理(2021年2期)2021-03-16
文萃报·周二版(2019年39期)2019-09-10
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
