
7-氨基-3-乙烯基头孢烷酸的一步法环保制备方法
2021-07-04 02:53徐鑫林梁丙辰姜鹏鹏
煤炭与化工 2021年5期
徐鑫林,梁丙辰,姜鹏鹏
(1.河北合佳医药科技集团股份有限公司,河北 石家庄052165;2.河北合佳医药科技集团合佳研究院,河北 石家庄050035)
0 引 言
7-氨基-3-乙烯基-4-头孢烷酸(7-AVCA)是头孢克肟和头孢地尼的药用中间体。头孢克肟和头孢地尼均为第3代头孢菌素,具有广谱抗菌、对β内酰胺酶稳定、肾毒性小等特点,在抗生素领域应用广泛。由于门诊输液受到限制,口服类头孢菌素的治疗效果表现良好。
目前,7-AVCA的合成路线主要有如下3种工艺路线。
(1)以青霉素G钾盐为原料,经酯化、开环、扩环、Wittig反应等系列反应合成。
(2)以7-氨基头孢烷酸(7-ACA)为原料,经硅烷化保护、碘代、无水条件下进行Wittig反应合成。
(3)以7-苯乙酰氨基-3氯甲基头孢烷酸对甲氧基苄酯(GCLE)为原料,经季膦化Wittig反应制得7-苯乙酰氨基-3乙烯基头孢烷酸对甲氧基苄酯(GVNE),GVNE脱除羧基和氨基保护基制得。
1 3种工艺路线的特点
路线(1):原料便宜易得,但步骤多、收率低、环境污染严重。
路线(2):反应条件苛刻、工业操作不宜控制,以上这2条路线都难以实现产业化。
路线(3):以GCLE为起始原料,反应条件温和,是目前我国已经工业化的合成路线。
由于GCLE的分子量比7-AVCA大很多,制备时会有2个较大的基团脱去,生成大量的副产物,使得原子利用率低,且质量收率差,导致了生产成本的增加。
另外,用GCLE制得中间体GVNE,再水解侧链生成7-AVCA,需经卤代、季膦化、乙烯基化、酸解、酶解等多种工序,工艺操作复杂,废水和废液的产生量大。
实验研究发现,对D-7-ACA进行分子改造以制备7-AVCA,其工艺过程具有所需的步骤少、原子利用率高、三废产生少等特点,是一种绿色环保的制备方法。
2 实验部分
2.1 实验主要设备及试剂
2.1.1 实验主要设备
(1)高效液相色谱仪:Agilent 1260 Infinity ii型,安捷伦科技有限公司生产。
(2)色谱柱:250 mm×4.6 mm,5μm,C18,大连依利特分析仪器有限公司生产。
(3)恒温搅拌器:S212-90型,上海申科仪器有限公司生产。
(4)pH计:FE28型,梅特勒-托利多集团生产。
(5)恒温水浴:HB10,艾卡(广州)仪器设备有限公司生产。
2.1.2 实验主要试剂
(1)D-7-ACA、7-AVCA均为工业级(浓度≥98%),由河北合佳医药科技集团股份有限公司提供。
(2)氯铬酸吡啶(P.C.C)、甲基三苯基溴翁盐、乙腈、甲醇、四甲基胍均为分析纯,为麦克林试剂。
(3)浓盐酸、乙酸钠、冰醋酸均为分析纯,由天津市永大化学试剂有限公司提供。
(4)高效液相色谱仪流动相所用溶剂乙腈为色谱纯,为麦克林试剂。
2.2 HPLC检测条件
(1)仪器:Agilent高效液相色谱仪。
(2)色谱柱:250 mm×4.6 mm,5μm,C18。
(3)流动相:A液:乙腈;B液∶5.0 g乙酸钠+1 000 mL水,乙酸调节pH=4.5;A液∶B液=30∶70。
(4)柱温:30℃。
(5)进样量:10μL。
(6)流速:0.5 mL/min。
(7)检测器波长:254 nm。
(8)洗脱时间:40 min。
2.3 7-AVCA的合成
(1)A液。
首先,将11.51 g(0.05 mol)D-7-ACA加入到250 mL四口瓶中,再加入80 mL乙腈和11.5 mL水,控温25℃。
然后,在四口瓶中加入6.33 g四甲基胍溶解,部分溶解后,再加入10.78 g氯铬酸吡啶(P.C.C),反应2 h。
最后,使用高效液相色谱法(HPLC)检测D-7-ACA残留(≤5%),留存备用。
(2)B液。
将35.72 g(0.1 mol)甲基三苯基溴化膦加入到250 mL四口瓶中,再加入100 mL乙腈,25℃条件下搅拌溶解,留存备用。
(3)合料。
在25℃条件下,将B液缓慢加入至A液中,滴加50 mL 1 mol/L碳酸钾溶液,调节溶液pH=7~9,反应1.5 h,使用HPLC检测A液中产物的残留(≤5%)。
反应完毕后,用1 mol/L盐酸调节溶液pH=4,用时2 h。降温,使温度<10℃。过滤,滤饼用60%甲醇水冲洗。在40℃条件下真空干燥,即得7.01 g白色固体,物质的量收率约为62.0%,纯度约为99.1%。
3 反应原理
D-7-ACA经弱氧化剂氯铬酸吡啶(P.C.C)氧化后,3号位羟甲基氧化为醛基。由于P.C.C氧化性较弱,不会对头孢环及7号位氨基产生破坏。醛基与甲基三苯基溴化膦在碱性条件下发生Wittig反应,生成乙烯基,制备出7-AVCA。
由D-7-ACA制备7-AVCA的反应过程如图1所示。
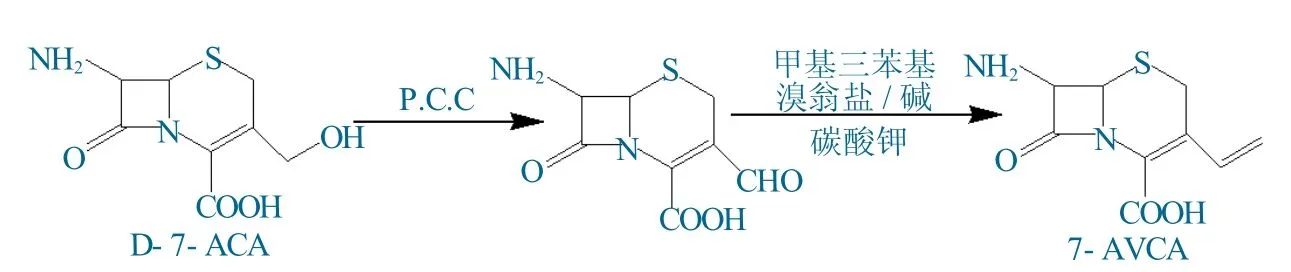
图1 D-7-ACA制备7-AVCA的反应过程Fig.1 Reaction process of preparing 7-AVCA from D-7-ACA
4 结果与讨论
4.1 高效液相色谱检测结果
D-7-ACA制备7-AVCA分为以下2步。
第1步:P.C.C氧化D-7-ACA的过程,其HPLC色谱如图2所示。
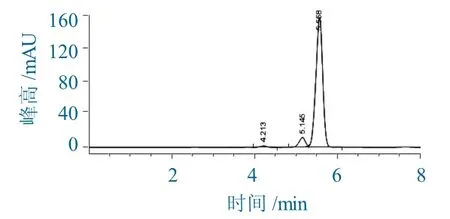
图2 P.C.C氧化D-7-ACA的HPLC色谱Fig.2 HPLCchromatography of the oxidation of D-7-ACA by P.C.C
由图2可以看出,5.145 min为D-7-ACA残留峰,5.551 min为P.C.C氧化产物。
第2步:氧化物经Wittig反应制备7-AVCA,其HPLC色谱如图3所示。
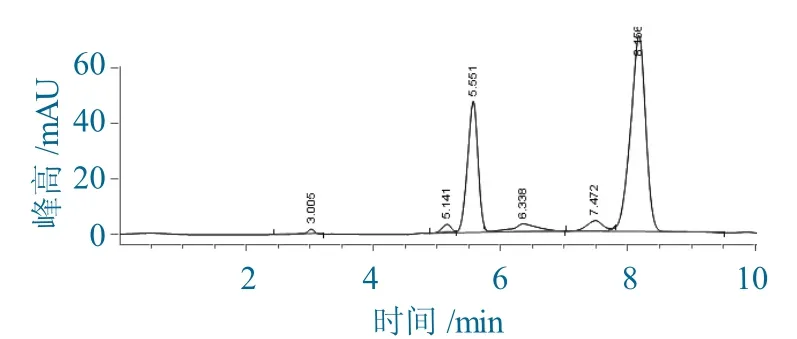
图3 Wi t t i g反应制备7-AVCA的HPLC色谱Fig.3 HPLCchromatography of 7-AVCA prepared by Wittig reaction
由图3可以看出,5.551 min为P.C.C氧化产物,8.156 min为产品7-AVCA。
4.2 有机碱种类对氧化反应时间的影响
实验发现,固态下D-7-ACA的3号位羟甲基氧化较难进行,而D-7-ACA在常规单一的混合有机溶剂中的溶解度较小,故加入有机碱与羧基成盐使其溶解。
D-7-ACA的溶解程度与其被氧化所需的时间成正比,为便于反应进行,需筛选较好的有机碱助溶剂。固定其它反应条件不变,添加相同量的不同有机碱,测定反应条件下D-7-ACA残留合格所需的时间。
有机碱种类与反应时间的关系见表1。

表1 有机碱种类与反应时间的关系Table 1 Relationship between organic base types and reaction times
由表1可以看出,反应时间与有机碱的碱性强弱有关,但并非碱性越强,反应越快。综合以上结果,其中效果最好的为四甲基胍。
4.3 甲基三苯基溴化膦加入量对产物收率的影响
为增加原料的转化率,采用增加另一反应原料的方法进行实验。实验发现,甲基三苯基溴化膦加入量对收率有较大影响。
固定D-7-ACA的投料量,在其它条件不变的情况下,改变甲基三苯基溴化膦的投料量,并成比例改变碳酸钾溶液的加入体积,甲基三苯基溴化膦与原料D-7-ACA的物质的量比与产物收率的关系如图4所示。
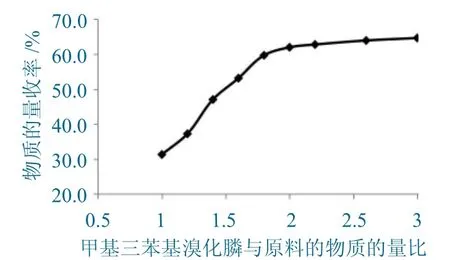
图4 甲基三苯基溴化膦与原料加入量的物质的量比与产物物质的量收率的关系Fig.4 Relationship between molar ratio of added amount of methyl triphenyl phosphonium bromide and rawmaterial and molar yield of product
由图4可以看出如下内容。
(1)等比例加入甲基三苯基溴化膦产物,收率极低,产物收率随甲基三苯基溴化膦加入比例的增加而增加。
(2)当甲基三苯基溴化膦增加至D-7-ACA投料量的2倍后,再增加甲基三苯基溴化膦的投料量,其产品的收率提高不明显。
综合环保和经济方面考虑,选择甲基三苯基溴化膦的投料量为D-7-ACA投料量的2倍物质的量比为最佳投料量。
5 结 论
通过改造D-7-ACA的分子结构,提出了一种全新的制备7-AVCA的一步法环保合成工艺。该工艺方式较原有的工艺方式操作简单,改变了目前通用的由GCLE路线制备7-AVCA原子利用率不高的问题。
D-7-ACA溶解后经P.C.C氧化,3号位醛基与甲基三苯基溴翁盐在碱性条件下进行Wittig反应,制备7-AVCA。
本方法给出了2个工艺的关键点。
(1)最优有机碱助溶剂为四甲基胍。
(2)甲基三苯基溴化磷投料量为D-7-ACA的2倍物质的量。
通过该工艺所制备出的产品7-AVCA,其质量与原工艺无异,同时避免了因进行酶水解反应而导致产品的发酵味道,整体原子利用率较高。所制备的产品7-AVCA物质的量收率约为62.0%,纯度约为99.1%。
猜你喜欢
玻璃(2022年1期)2022-02-23
中国烟草学报(2021年4期)2021-09-26
水泵技术(2021年4期)2021-01-22
农药科学与管理(2019年8期)2019-11-23
山东煤炭科技(2018年1期)2018-12-05
乐活老年(2016年10期)2016-02-28
化学工业与工程(2015年1期)2015-02-10
中国药业(2014年12期)2014-06-06
中国合理用药探索(2014年11期)2014-03-11
无机化学学报(2014年12期)2014-02-28
