
不同形貌金属氧化物的制备及其在工业催化反应中的应用
2021-06-30 01:27周石杰任祯杨宇森卫敏
化工学报 2021年6期
周石杰,任祯,杨宇森,卫敏
(北京化工大学化学学院,化工资源有效利用国家重点实验室,北京100029)
引 言
金属氧化物催化剂是一类重要的工业催化剂,其在逆水煤气变换、CO氧化、选择性加氢等多种重要的工业反应中发挥着不可替代的作用,引起了国内外研究者的广泛关注。然而,金属氧化物通常使用多步制备方法:在水相中沉淀金属离子以形成金属氢氧化物,然后对氢氧化物进行热处理以生成氧化物,极快的沉淀速度阻碍了对氢氧化物前体大小和形貌的精确控制,而通常在高温下发生的脱水过程并不是拓扑转变,因此氢氧化物和氧化物之间的晶体结构差异很大。此外,在脱水过程中除去水会产生大量无序的孔和表面空位,从而导致表面粗糙和多晶的形成。另一方面,金属氧化物通常应用于高温下的化学反应中,并且它们的表面空位和晶格氧物种经常参与催化循环,导致尺寸和形貌的显著变化,在催化反应过程中温度和反应气氛促使氧化物表面重构为热力学更稳定的表面,从而使总表面能减至最小[1]。晶体学结构的这些不确定性使得构效关系的理解变得困难。随着人们对工业生产的要求不断提高,制备形貌可控的金属氧化物催化剂引起科研工作者的广泛关注。
金属氧化物[2-4]是一类无机材料,在传感器、催化剂、燃料电池等领域具有广泛应用。氧化物表面包含不同类型的缺陷(纽结、台阶、平台),可能在催化反应中起作用,例如导致金属氧化物催化剂对催化反应的结构敏感性。近年来,随着纳米材料突飞猛进的发展,金属氧化物由于自身结构具有独特性,例如酸性(Lewis和Brønsted型)、碱性、氧化还原特性(当存在过渡金属离子时)[5-8],在氧化(选择性氧化或完全氧化)、酸碱催化、光电催化和生物质转化等诸多领域表现出广泛的应用前景[9-16]。金属氧化物催化剂的性能在很大程度上取决于它们的形貌和结构,纳米材料的形貌是其结构的微观反映,不同形貌的金属氧化物会直接影响催化剂的整体性能。对于各种复杂形貌的金属氧化物纳米晶体的制备,形貌可控的晶体生长受动力学控制,其中低能晶面持续存在,而高能晶面消失,因此在合成体系中添加表面活性剂和模板剂是制备具有不同形貌的金属氧化物的常用方法,因为表面活性剂可以优先吸附在某些特定的晶体平面上,然后改变纳米晶体的生长方向和速率[17-22]。本文总结了不同形貌的金属氧化物的制备方法、生长机制及其结构特性,特别地,聚焦于金属氧化物催化剂在氧化反应、加氢反应中的最新研究进展。详细讨论了催化剂活性结构的表征、确定催化性能与催化剂形貌的内在关系,为金属氧化物催化剂的合理设计以及制备提供了一定的理论依据。希望这篇综述能够吸引更多研究者关注金属氧化物催化剂,鼓励研究者们在这一迅速发展的领域开展富有创新性的工作。
1 不同形貌的金属氧化物的结构特性
近年来,材料科学的飞速发展为调控金属氧化物的形貌提供了更大的可行性,特别地,在纳米水平上对金属氧化物的形貌控制可以选择性地暴露反应性晶面,从而可以极大地提高它们的催化性能[23-30]。除了金属氧化物的固有特性外,金属氧化物优势暴露的晶面是影响一系列反应的关键因素,例如CO2甲烷化[31-32]、CO氧化[33]、逆水煤气变换(RWGS)[34]、甲烷燃烧[35]。金属氧化物晶面对催化性能的影响的差异起源于表面原子排列和密度差异,即晶面效应,这影响了氧空位形成能,而在这些反应中,催化活性与氧空位的数量密切相关,对于不同形貌的金属氧化物的晶面效应,前人已经进行广泛的研究。Xie等[36]报道了一种以(110)为主要暴露晶面的Co3O4纳米线作为CO氧化反应催化剂,而(110)作为优势暴露晶面极大地促进了CO的吸附,吸附上的CO可以与邻位O(与周围三个Co3+配位)发生反应生成CO2,并把Co3+还原成Co2+,生成的Co2+又可以用于O2的活化。Tian等[37]制备了棒状、片状和立方体状的Co3O4(分别表示为Co3O4-R,Co3O4-S和Co3O4-C),其优势暴露晶面分别为(110)、(111)和(100)面。程序升温还原(TPR)结果表明催化剂中氧的迁移率顺序为Co3O4-R>Co3O4-S>Co3O4-C,O2-TPD(氧气程序升温脱附)结果表明Co3O4-R具有最活泼的亲电子氧物种。生成活性氧物种是催化氧化反应的关键步骤,理论计算(DFT)表明Co3O4-R在(110)面上的氧空位形成能最低,由于其具有最大的O2吸附能而具有较高的氧活化能力,Co3O4-R对于丙烷氧化反应具有出色的活性,仅在195℃下丙烷的转化率就达到90%。此外结合二氧化碳程序升温脱附(CO2-TPD)、氨气程序升温脱附(NH3-TPD)以及原位漫反射红外光谱(in situDRIFTS)的结果证实了Co3O4-R中存在的最大量的Lewis酸-碱对极化使底物电子分布极化,从而加速了C—H键的活化,亲电氧物种(O22-或O-)导致碳骨架降解,并在矿化为CO2之前形成了羧酸盐中间体。Tumuluri等[38]通过原位红外光谱(in situFT-IR)和质谱(MS)技术以及DFT计算探究了TiO2纳米棒{(010)+(101)+(001)},圆盘{(001)+(101)}和截断八面体{(101)+(001)}三种纳米晶体的表面结构对CO2吸附模式、数量和强度以及抗SO2性能的影响。吸附CO2后,在TiO2棒和圆盘上会形成羧酸盐和碳酸盐(桥连的单齿碳酸盐),而在截短的TiO2八面体上只会形成双齿碳酸盐和单齿碳酸盐。通常CO2吸附物种的热稳定性顺序为:羧酸盐≈单齿碳酸盐>桥接碳酸盐>双齿碳酸盐≈重碳酸盐,实验上也通过TPD证实了TiO2纳米棒和圆盘比截短的TiO2八面体对于CO2的吸附更强,这归因于与截短的八面体相比,纳米棒和圆盘上低配位数的表面氧和氧空位数量更多。红外研究表明CO2吸附模式和强度受SO2的影响。在三种TiO2纳米晶体中,由于在SO2吸附循环中形成的硫酸盐积累,截短八面体的CO2吸附强度下降程度最大。Tawfilas等[39]报道了利用1H时域核磁共振氢谱(1H-TD-NMR)区分锐钛矿、板钛矿和金红石型TiO2晶体结构,利用水分子与TiO2纳米晶体之间的相互作用,可以将质子横向弛豫时间(T2)与浓度和比表面积(δp×Cm)的函数相关联,并作为晶相的指标。弛豫度(R2)与浓度(Cm)之间呈线性关系,在对特定表面进行归一化之后,所得的斜率表示表面/水的相互作用,并且范围从金红石纳米晶体的1.28 g/(m2·s)到具有相似尺寸的板钛矿的0.52 g/(m2·s),根据表面能的趋势,更高的斜率[1.85 g/(m2·s)]对应于较小的金红石型纳米晶体。NMR技术对诸如羟基化之类的表面处理也很敏感,因此通过结合溶剂弛豫确定形貌的其他已建立的方法,通过时域核磁共振(TD-NMR)技术,利用水作为探针来识别同一氧化物和晶体的不同相,不同的比表面积并获得有关表面结构的详细信息。Bao等[40]利用包括优势暴露晶面为(100)面的Cu2O立方体、(111)面的Cu2O八面体和(110)面的Cu2O菱形十二面体作为模型催化剂,研究了晶面的表面化学性质对于CO氧化反应催化性能的影响。催化性能遵循八面体菱形>十二面体>立方体的顺序,表明Cu2O(111)晶面在CO氧化中活性最高。一氧化碳程序升温还原(CO-TPR)表明Cu2O八面体是其中最容易还原的。DRIFTS表明CO在Cu2O纳米晶体上的化学吸附取决于其Cu2O形状和化学吸附温度。在菱形十二面体上,308℃时CO化学吸附最强,而150℃时则为八面体上CO化学吸附最强,因此三种Cu2O纳米晶体对CO的还原性和化学吸附能力与其在CO氧化反应中的催化性能一致。Cu2O纳米晶体的表面化学性质和在CO氧化反应中的催化性能与其暴露的晶面和表面组成/结构相关,Cu2O八面体使(111)晶面具有配位不饱和CuⅠ位点,因此在化学吸附CO和催化CO氧化方面活性最高。Hua等[41]合成了八面体Cu2O(o-Cu2O)和立方体Cu2O(c-Cu2O)纳米晶体,通过改变纳米颗粒的形貌和暴露晶面控制纳米颗粒的表面重整过程以及重整表面的结构和催化性能。结合扫描电子显微镜(SEM)、X射线光电子能谱(XPS)和氢气程序升温还原(H2-TPR)等表征证实了CuO/o-Cu2O和CuO/c-Cu2O具有不同的表面结构。DFT计算证实了Cu2O(111)的最佳表面结构不同于Cu2O(100)的表面结构,配位不饱和铜原子存在于Cu2O(111)面上,但不存在于Cu2O(100)面上,而Cu2O的优势暴露晶面控制CuO覆盖层的表面结构,Cu2O(111)面上的CuO覆盖层端位为3配位的Cu(Cu3c)和3配位的氧(O3c)原子,而Cu2O(100)面上的CuO覆盖层端位为O2c原子,CuO/o-Cu2O催化的CO氧化表观活化能比CuO/c-Cu2O催化的表观活化能低。
甲醇被认为是用于研究氧化物表面性质的有效探针分子,Wu等[42]利用红外光谱探究了棒状、立方体和八面体的CeO2在甲醇吸附和反应过程中表面缺陷类型及原子配位数的变化,发现端式吸附的甲氧基存在于棒状、立方体、八面体三种形貌的CeO2纳米晶体上;而采用桥式和3配位模式吸附的甲氧基仅存在于立方体和八面体形貌的CeO2上。结合紫外拉曼光谱(UV Raman spectroscopy)表征结果,CeO2表面的本征缺陷浓度遵循如下顺序变化:八面体<立方体<棒,因此CeO2其表面缺陷类型和浓度与形貌密切相关。以CO的红外吸附作为探针,能够获得不同晶面下CO振动峰的确切归属,为不同形貌的氧化铈纳米粒子的确切的表面提供了具体的原子结构信息,此外,从纳米粒子到单晶表面都可以得到较好的红外振动信号,可以充分联结两者间“材料鸿沟”的桥梁。Yang等[26]通过红外光谱探究了实际情况下CeO2纳米棒的晶面暴露,研究发现纳米棒所暴露的(110)晶面由于表面重构现象的大量发生,会转化为具有低能稳定的(111)面。以CO吸附的红外吸收光谱获得了在CeO2(111)和CeO2(110)晶面上C—O不同的伸缩振动峰。根据不同暴露晶面上氧化还原程度的差异,相对于(111)晶面,在(110)晶面对应的氧化态和还原态的CO振动峰的位置向更高波数偏移,同时还可能伴有表面Ce3+物种附近的CO吸附特征峰以肩峰的形式存在。随后对CeO2进行溅射和退火处理,再通过CO的吸附进行表面氧化还原位点的研究,经过溅射和退火处理后的CeO2的(110)晶面通过CO的吸附进行表面氧化还原位点的探测同时显示(110)和(111)晶面的CO特征振动峰,并且在(111)表面出现更强的振动峰,说明单晶CeO2(110)表面发生了明显的重构现象,暴露的(110)经过表面重构为(111)。同时结合TPSR(程序升温表面反应)表明从60 K到100 K的逐渐升温过程,(111)面的CO更容易扩散到(110)表面,从而增加(110)面上的CO的吸附峰强度。除了优异的氧化还原性之外,金属氧化物独特的酸碱性质也因其作用而受到人们的广泛关注。Vilé等[27]通过控制合成条件得到具有特定表面结构和组成的棒状、立方体、八面体等不同形貌的纳米CeO2,通过与各种探针分子(吡啶、乙腈、CO2和氯仿)结合的原位红外光谱,研究了酸碱位对二氧化铈形貌的表面结构依赖性,研究发现三种形貌的二氧化铈上均存在较弱且强度相似的Lewis酸位点。CO2吸附结果表明二氧化铈上存在两种类型的碱性位点,即羟基和表面晶格氧。CO2和氯仿的吸附表明Lewis碱的强度和数量与形状有关,即:棒状>立方体>八面体,Ce阳离子和O阴离子配位不饱和状态的表面结构与三种形貌的二氧化铈上缺陷位点数量之间相互作用导致CeO2具有弱表面依赖性和强表面依赖性的酸碱位点。除此之外,表面的Ce具有不同的电子态密度(Lewis酸)与不同晶面的表面能有关,影响反应物的吸附状态和活化形式。受限于目前常规的表面表征手段,对不同晶面上Ce的化学状态的认识仍存在争议和不足,不能对酸碱位点的数量和强度得到确切的认识。Tan等[30]成功地合成了不同晶面暴露的球形、八面体、棒状以及立方体形貌的CeO2纳米粒子,利用三甲基膦(TMP)作为探针分子,借助了31P固体核磁(TMP-31P-NMR)的表征手段得到了Ce的化学状态和材料形貌的密切联系。根据TMP-Ce处的31P化学位移与形貌存在的对应关系[-33(八面体)>-40.5(球体)>-47.5(棒状)>-58(立方体)],结合电镜手段观测到不同形貌的CeO2不同的优先暴露晶面,因此建立起固体核磁的化学位移和不同形貌暴露晶面的联系。CeO2表面Ce物种的酸度(或化学状态)依赖于其暴露的晶面,因此Ce在(111)面上具有较强的Lewis酸强度(更多的Ce4+),在(100)面上具有较低的Lewis酸强度(更多的Ce3+)。建立的CeO2的TMP-31P-NMR指纹图谱可以反映给定CeO2样品的晶面分布和浓度,CeO2不同晶面上的Ce化学状态的变化,反映了由于不同的Ce配位环境引起了电子密度的“连续”变化,为不同暴露晶面和不同形貌氧化物的晶面分布和浓度研究提供了一个有效的新途径。
虽然研究者采用实验方法对不同形貌的金属氧化物的晶面以及晶体参数等进行了研究,但是难以获得微观结构和电子结构等方面的信息;而DFT计算能够从微观原子级别很大程度弥补实验上的不足。Zhang等[43]采用DFT计算研究了CeO2的(100)、(110)和(111)晶面上解离H2和氧空位的生成。研究发现CeO2(100),(110)和(111)表面上H2相比于均裂,异裂更容易发生,并且CeO2(100)表面表现出最佳的H2活化性能。在整个催化反应过程中H2作为氢源用以构筑有缺陷的表面,氧空位处的电荷损失促进CeO2(100)表面生成甲酸盐途径中甲醇合成的关键中间体bi-H2CO*。Hua等[25]制备了Cu2O八面体、立方体和菱形十二面体用于探究催化丙烯氧化中的晶面控制选择性,结合C3H6和C3H6+O2化学吸附的漫反射傅里叶变换红外光谱(DRIFTS)、C3D6化学吸附的DRIFTS以及DFT计算发现优势暴露晶面为(111)的Cu2O八面体对形成丙烯醛的选择性最高,1配位的Cu+是活性位点。优势暴露晶面为(100)面的Cu2O立方体对形成CO2的选择性最高,2配位的O是活性位点。优势暴露晶面为(110)面的Cu2O菱形十二面体对环氧丙烷的选择性最高,3配位的O是活性位点。Huang等[29]通过in situDRIFTS光谱探测CO和CO2化学吸附来探究CeO2、TiO2和Cu2O纳米晶体不同晶面的表面结构。以CeO2为例,八面体CeO2上CO的化学吸附主要以1579和1296 cm-1处的ν(CO3)双齿碳酸盐和1620 cm-1处的ν(CO3)碳酸氢盐模式存在,而在立方体CeO2上形成了多种表面物质,包括桥式碳酸盐[在1395和1218 cm-1处的ν(CO3)],双齿碳酸盐,多齿碳酸盐{在1470 cm-1处的多齿碳酸盐ν(CO3),碳酸盐/甲酸盐[ν(OCO)在1326 cm-1],碳酸氢盐[ν(CO3)在1605 cm-1]和碳酸盐[ν(OCO)在1277 cm-1}。在CO化学吸附作用下,八面体CeO2和立方体CeO2上形成的表面物质之间的差异与CeO2(111)和(100)晶面的表面结构有关。CeO2(111)表面最外层的相邻O3C原子均通过第二层上相同的Ce7C原子连接,因此只能形成双齿碳酸盐物种,而桥连碳酸盐物种不能形成。CeO2(100)最外层的相邻O2C原子是通过第二层上相同的Ce6C原子连接的表面,因此双齿碳酸盐和桥式碳酸盐都可能形成。CO在立方体CeO2上的化学吸附过程中还观察到表面羟基,这可能是由于残留的CO在表面氧空位上解离,因此CO化学吸附可以探测具有不同形貌的CeO2纳米晶体上暴露晶面的不同表面结构。化学吸附在金属阳离子位点的CO或CO2以及涉及O位点的双齿碳酸盐和桥式碳酸盐对表面组成以及表面金属阳离子和O阴离子的局部配位环境敏感,并且可以与氧化物纳米晶体暴露晶面的表面结构很好地关联。通过CO化学吸附形成的碳酸盐和碳酸盐物种可以探测CeO2的不同晶面。由CO化学吸附形成的碳酸盐物种可以探测TiO2的不同晶面。CO化学吸附形成的吸附的CO和碳酸盐物种可以探测Cu2O的不同晶面,而CO2化学吸附形成的吸附的CO2也可以探测Cu2O的不同晶面。Wang等[44]采用DFT计算探究了Co3O4(001)和Co3O4(011)表面上的晶格氧离子进行CO氧化反应,CO氧化反应对晶面和活性位点表现出显著依赖性,表面Co-O离子对是CO氧化的活性位点。作为CO氧化反应活性的指标,CO2形成的过渡态能级顺序为(011)-Co-Ot≫(001)-Co-Oo>(011)-Co-Oo>(001)-Co-Ot(Ot和Oo代表不同类型的氧,在晶格中有不同键合方式。Ot表示与1个Co2+和1~3个Co3+键合,Oo表示附近所有离子均为Co3+),因此(011)面上的Co-Ot位点表现出最高的CO氧化活性。低配位的表面离子有助于提高CO氧化活性,(011)面上的4配位的Co3+离子和2配位的Ot离子比(001)表面上的5配位的Co3+离子和3配位的Ot离子活性更高,而过渡态能级和氧空位形成能正相关,因此氧空位形成能最低的Co3O4(011)面的Co-Ot离子是CO氧化反应中活性最高的位点。
在多相催化领域中,金属氧化物催化是最重要的领域之一,单金属氧化物、混合金属氧化物、尖晶石型氧化物和钙钛矿型氧化物等金属氧化物催化剂被广泛应用于工业生产过程。其中与不可还原性单金属氧化物相比,可还原性单金属氧化物由于金属的多价态特性可以在不同高低价态间形成一个氧化还原的循环反应,在金属价态改变的过程中通过晶格氧的释放和氧空位的形成可以产生大量的表面吸附氧物种,因此在工业催化领域具有广泛应用。对单金属氧化物进行第二或多个金属的掺杂形成的混合金属氧化物有助于提高催化剂的反应活性和热稳定性。尖晶石型金属氧化物同时具有多个价态可以同时存在A位和B位上,是一种价格低廉、资源丰富、环境友好并且可以大规模生产的优良催化剂。钙钛矿型氧化物具有优良的结构稳定性,电子转移和氧化还原性能,在催化领域被广泛研究。有序孔结构的钙钛矿具有大比表面积,不仅有效增加催化剂的活性位点,还可以作为其他活性成分的载体,提供充足的内外比表面积和热稳定性能。已有大量不同形貌的金属氧化物研究,然而将具有特定形貌的金属氧化物材料规模性生产并工业化仍旧存在挑战。主要原因在于制备方法复杂以及相关的高成本问题,此外金属氧化物其制备方法和功能-结构关联仍需作更多的研究,以功能为导向的研究在催化应用上具有相当大的研究潜能。
2 不同形貌的金属氧化物的制备
2.1 模板法制备不同形貌的金属氧化物
微波辅助[45]、电沉积法[46]、化学气相沉积[47]作为制备不同形貌的金属氧化物的常用方法,存在反应时间长、步骤烦琐及仪器设备昂贵等诸多方面的缺点,阻碍了其在工业上的应用与发展。而液相法具有低温、操作简便、大规模合成等优点,但是反应时间、反应温度、反应物比例以及表面活性剂和模板的加入等诸多影响因素使得不同形貌金属氧化物形成过程及形成机理的研究变得困难,本文从是否添加表面活性剂或模板对液相法制备出来的几种不同形貌的典型金属氧化物的形成过程以及形貌演变机制进行了总结。Xiao等[48]以乙二醇和葡萄糖作为表面活性剂,Co(NO3)2·6H2O作为钴源,通过溶剂热法合成了氢氧化钴碳酸盐水合物,随后通过焙烧首次成功大规模制备出具有海胆状结构的3D分层Co3O4双球(图1)。前驱物的形貌决定最终产物的结构,随着反应时间增加,从纳米棒演变成束状结构,再到花叶状结构,再到哑铃状颗粒,最后变成双球体。基于时间对前体形貌的影响,他们提出了一种多步分解生长机制来解释3D分层双球体的形成:(1)在反应体系中成核后,前体快速生长产生纳米棒。(2)由于随后的分裂,生长可以发生在纳米棒的两个头部和分支的纳米棒上。(3)分裂生长不仅可以连续地从纳米棒中发生,而且可以从由捆状束组成的纳米针新生成的尖端发生。每代中长度约为1.5μm的纳米棒可以分裂成两个以上直径较小的纳米针,在第一代或第二代分裂生长中可以产生具有束状结构的前体。(4)由于生长可以在纳米针的尖端反复进行,因此分裂前三个后,前体纳米针的数量爆炸式增加。由于新产生的纳米针的方向始终与一维纳米针晶种的轴相关并有偏差,因此最终可以从纳米棒的多步分裂生长中产生两个半球,从而形成3D分层的哑铃状结构。

图1 在160℃下前体氧化物合成的FE-SEM图:2 h(a)、3 h(b)、4 h(c)、5 h(d)、6 h(e)[48]Fig.1 FE-SEM images of the precursors synthesized at 160℃for 2 h(a),3 h(b),4 h(c),5 h(d),6 h(e)[48]
Wang等[49]通过简便的水热法合成了长的氢氧化钴碳酸盐水合物Co(CO3)0.5(OH)·0.11H2O前体纳米线,随后通过焙烧获得了具有较大长宽比的多孔Co3O4纳米线。添加剂(尿素)的量对合成后的氢氧化钴碳酸盐水合物中间体的形貌有重要影响,可通过改变尿素浓度来控制其均一性和整体结构。尿素的缓慢水解提供的OH-和CO32-可能会有利于纳米线沿着其纵轴逐渐生长,尿素浓度较低时得到分散性较好的纳米线,尿素浓度升高导致纳米线黏连成束状,当浓度进一步升高将会得到直径为5μm的纳米线向四周辐射的海胆状微米球。他们认为通过尿素水解产生的碳酸根离子不仅参与中间体化合物的形成而且促进纳米线的各向异性生长,而且还表现出类似于配位剂的作用,有助于在较高的浓度下构筑更复杂的层次结构。Wang等[50]采用金属有机骨架(MOF)作为模板并使用次序模板法(STA)合成了Co3O4十二面体的多壳层中空结构(HoMS),由于MOF中金属原子的拓扑排列,HoMS中的Co3O4纳米晶体沿所需的方向组装,形成主要暴露晶面为(111)的独特壳层。通过调节程序升温速率和氧气分压可以设计出具有不同壳层数的HoMS,通过将700~800 nm和2.5μm ZIF-67在空气中于425℃下以0.5℃/min的升温速率焙烧1 h可以获得DS(DS代表2壳层)和TS(TS代表3壳层)Co3O4HoMS,通过2.5 μm ZIF-67在气体条件下(O2∶N2=1∶9)在425℃下以0.5℃/min的升温速率焙烧1 h可以获得QS(QS代表4壳层)Co3O4HoMS。ZIF-67中暴露的(001)和(011)面显示出周期性排列的Co原子,类似于Co3O4上(111)面的Co原子排列,ZIF-67仅需要简单地移动Co原子即可进行演变。此外MOF中的有机配体和孔为位移和重排提供了足够的空间,Co3O4HoMS与ZIF-67的十二面体结构相同,表明从MOF到Co3O4HoMS的形貌继承,而且从HoMS的外侧到内侧所有壳层都具有相同的十二面体结构。Yin等[51]通过控制焙烧钴(Ⅱ)复杂前体的升温速率制备具有可控壳层数的均匀Co3O4中空球以及Co3O4蛋 黄-蛋壳结构。形成具有不同中空结构的Co3O4涉及两个步骤:(1)通过六水合硝酸钴(Ⅱ),乙酰丙酮和水合肼在80℃下反应制备Co基前体。(2)对Co基前体进行热处理,以在可控的加热速率下获得不同的Co3O4空心结构。对于在10℃/min退火的样品,其蛋黄-蛋壳结构的内部核直径约为750 nm,核与壳之间的间隙为10~20 nm。对于在5℃/min退火的样品,表面变粗糙且破裂的球体表明其为空心结构,空心微球的壳层数2,壳的厚度约为25 nm。形成不同的中空结构的机制可能是由于内层和外层的不同生长速率引起的。前体球外部存在足够的氧气而在前体球内部没有足够的氧气,外层中Co3O4晶体的生长速度快于内部,当加热速率增加时,在球体表面迅速形成Co3O4,并且内部反应剧烈并同时收缩,形成核壳结构,当加热速率降低时,则微球内部的反应速率将减慢,因此将形成壳层数不同的Co3O4中空球。
Jiang等[52]通过使用甘氨酸作为软生物模板简单沉淀成功合成了具有各种形貌的3D多孔CeO2,CeO2的形貌取决于反应物的摩尔比,在不引入甘氨酸的情况下获得了具有约10 mm纺锤形结构和少量球形颗粒的A-CeO2。随着甘氨酸的增加,球形颗粒消失,甘氨酸的摩尔比进一步增加,A-CeO2的形貌变为蝴蝶结和块状。甘氨酸的存在使A-CeO2的结构更加均匀,甘氨酸的含量影响了A-CeO2的形貌。此外尿素和Ce前体的量也会影响A-CeO2的结构,A-CeO2(1.5∶2∶1)呈棱柱形,A-CeO2(6∶2∶1)呈小哑铃形,A-CeO2(3∶2∶2)呈哑铃形。Xu等[53]通过简单的水热法制备了具有扫帚状多孔分层结构的CeO2。根据电镜表征结果,他们揭示形成扫帚状多孔CeO2的机理。首先溶液中通过结晶形成了许多纳米晶体,随着反应过程的进行,这些纳米晶体在柠檬酸钠的帮助下沿纳米棒的方向自组装,由于聚集的纳米棒之间的大间隙,这种聚集导致花状形貌的形成,随着Ostwald熟化过程的进行,许多纳米晶体在纳米棒表面形成并自组装,外部纳米粒子变大,而内部纳米粒子变小,最后由于纳米棒内部和外部之间的晶体浓度差异而消失,形成具有多孔结构的纳米棒。同时,纳米棒之间的间隙逐渐缩小,棒彼此靠近,在该阶段形成扫帚状的多孔结构。Li等[54]通过调节水相溶液的pH,使用简便的水热法合成了纳米晶体自组装的平均边长分别为562、353、65、52、46 nm的CeO2八面体。在pH为3时,形成平均边长为(562±54)nm的准八面体。随着前体溶液的pH增加到4,主要的产物是(353±47)nm的分级准八面体。前体溶液的pH变化导致纳米级的八面体具有尖锐的边角。在pH为5、6和7时,得到了明确的八面体,平均长度分别为(65±7)nm、(52±6)nm和(46±4)nm。他们提出了大小可调的分级CeO2八面体的可能形成机理:在水热条件下在前体溶液中形成CeO2初级纳米晶体,并在PVP的辅助下自组装成八面体聚集体。聚集体的粒径取决于初级纳米晶体的相对成核速率和自组装,通过控制前体溶液的pH,可以调节初级纳米晶体的成核速率。当前体溶液的pH低时,CeO2初级纳米晶体的成核速率缓慢,并且自组装占主导,这导致形成具有较大尺寸的分级八面体。前体溶液的pH增加会加速CeO2初级纳米晶体的成核,而自组装速率是恒定的,从而导致形成小尺寸的分层八面体。
Que等[55]通过简便的水热法基于明确的高结晶度纳米片合成了花状3D TiO2微结构。他们通过SEM观察锐钛矿型TiO2纳米片组装而成的花状3D分层结构的形貌演变,在反应初期(4 h)形成了大量具有不规则形状的薄片。随着反应时间延长至8 h,形状和尺寸没有明显变化,主要产物仍是分散的纳米片,但有些片开始通过交错和相互重叠而组装成交叉结构。将反应时间进一步延长至12 h,形成了越来越多的交叉结构。当反应时间控制在16 h,逐渐出现了由纳米片亚组装而成的花状结构,最后当反应时间延长至24 h,便获得了具有均匀而完美形态的花状TiO2微结构。基于以上对形貌变化的研究,他们微结构结构的形成归因于Ostwald熟化过程:(1)形成纳米片。(2)溶解一些纳米片,在残留的片上重结晶和生长。(3)花状层次结构的形成与生长。最初,TiO2纳米片是通过所谓的成核和随后的生长过程形成的,从热力学的观点来看,具有两个暴露晶面的单个片具有高表面自由能,为了降低总表面能,根据Ostwald熟化理论一些具有相对高得多的表面能的纳米片将溶解,然后再结晶并重新沉积在某处,最终获得了由TiO2纳米片组装的3D花状纳米结构。Pan等[56]发展了一种大规模合成均匀的、由一维单晶锐钛矿型纳米刺组成的海胆状介孔TiO2空心球(UMTHS)的新型策略,该方法涉及对自组装的非晶态含水TiO2固体球体(AHTSS)进行有针对性的蚀刻。在温和的水热条件下,在存在聚乙烯吡咯烷酮(PVP)表面涂层的情况下,自发重建表面氟化AHTSS形成UMTHS,调整实验参数实现UMTHS的可控合成,其球体尺寸可调,内部结构多样,包括实心、空心、核壳和蛋黄结构。他们根据实验现象推测了UMTHS的形成机理以及氟化物在结构演变中起到的作用:在PVP的保护下水作为唯一反应介质,较高的极性有助于TiO2在相对较低的温度110℃下结晶,反应温度和系统压力的显著降低有利于保持空心球的形态并控制锐钛矿型TiO2的晶体生长。氟化物的存在对于形成中空结构和海胆状的壳至关重要,随着F/Ti摩尔比(RF)的增加,由矿化剂氟化物诱导的晶体生长增强,在水热条件下,氟化物可以桥接TiO6八面体缩聚反应,从而使AHTSS逐步脱水并转变为锐钛矿相,并沿(101)平面优先生长。随着RF的增加,可以观察到从固体到空心球的清晰形态演变,以及从空心球分解的分离的纳米刺。可以以0.08的低RF获得由紧密堆积的纳米晶粒组成的介孔TiO2固体球(MTSS),将RF增加到0.12会增强蚀刻过程,从而在UMTHS中创建空心结构,同时由于氟化物引导的优先晶体生长,TiO2逐渐演变为纳米刺,但是将RF进一步增加到0.16会导致强烈的蚀刻过程并形成生长过度的纳米刺。因此在结构应力增加的情况下,只有少数空心球存在,而大多数空心球变形并坍塌成长纳米刺的碎片。
Dong等[57]通过水热条件下糖的乳液聚合反应制备碳质微球作为模板,通过简单的程序升温过程制备了具有明确结构的ZnO空心微球,不仅可以控制壳的数量,而且可以控制壳间距。通过程序升温过程实现多壳ZnO空心微球的结构演变(图2):(1)所有微球的直径相同,均为约1.0μm。(2)随着Zn前体浓度从3 mol/L增加到5 mol/L,壳数从2个(样品Ⅰ、Ⅱ和Ⅲ)增加到3个(样品Ⅳ、Ⅴ和Ⅵ)和4个(样品Ⅶ、Ⅷ和Ⅸ)。(3)以2℃/min(样品Ⅰ、Ⅳ和Ⅶ,快速加热)或1℃/min(样品Ⅱ、Ⅴ和Ⅷ,中等加热)加热到500℃时,产品为空心ZnO微球,其中同一微球中相邻壳之间的相距几乎相同。壳与微球中心的几何偏差是由壳在中空结构中的自由运动引起的。当以1℃/min的速度加热并在400℃保持30 min时,样品的空心微球的外部具有独特的密闭双壳结构(壳间距约为0.04μm)(样品Ⅲ、Ⅵ和Ⅸ,缓慢加热),壳没有碳残留物。通过分析图2中的演化过程,可以得出多壳ZnO空心微球的制备方案:较高浓度的锌前体会更深地渗透到碳质微球的核中,从而使其核内部存储更多的锌离子,并产生更多的内壳,从而形成四壳空心球(样品Ⅶ、Ⅷ),甚至带有更多壳的空心球,通过控制加热过程和前驱物的浓度,可以合成大量具有可控壳层数和壳间距的ZnO空心微球。
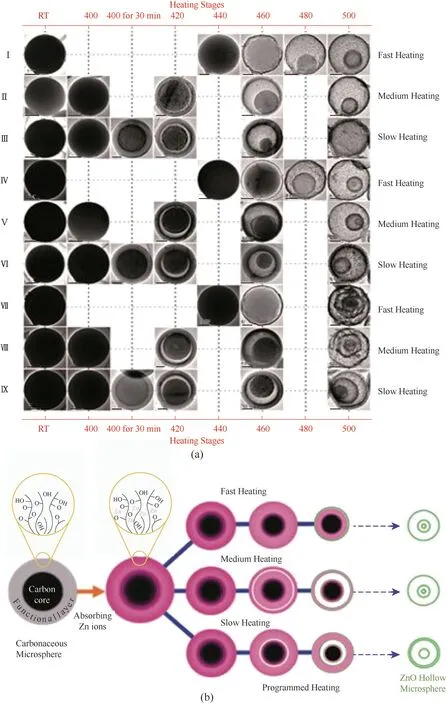
图2 多壳ZnO空心微球的演化过程。碳微球在加热之前(室温)和在不同温度(400℃、400℃下30 min、420℃、440℃、460℃、480℃和500℃)下加热后浸入硝酸锌溶液中的透射电子显微镜图像。3 mol/L和5 mol/L硝酸锌溶液分别用于样品Ⅰ、Ⅱ、Ⅲ和样品Ⅳ、Ⅴ、Ⅵ、Ⅶ、Ⅷ、Ⅸ。样品Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ和Ⅵ中使用的碳质微球的直径为3μm,样品Ⅶ、Ⅷ和Ⅸ中使用的碳质微球的直径为4μm。快速加热模式和中速加热模式下的温度分别在2和1℃/min下直接升高到500℃而慢速加热模式下的温度在1℃/min下直接升高到500℃并且在400℃保持30 min。在第一列中,样品Ⅰ~Ⅵ比例尺为1μm,样品Ⅶ~Ⅸ比例尺为1.3μm。第二列中的所有样品比例尺均为0.5μm,而第三列及更高列中样品比例尺均为0.3μm(a)。通过不同的加热过程形成多壳ZnO中空微球的图示(b)[57]Fig.2 Evolution process of the family of multishelled ZnOhollow microspheres.Transmission electron microscopy images of carbonaceous microspheresafter immersion in zinc nitrate solutions before heating(room temperature)and after heating at different temperatures(400℃,400℃for 30 min,420℃,440℃,460℃,480℃and 500℃).3 and 5 mol/L zinc nitrate solutions were used in samplesⅠ,Ⅱ,Ⅲand samplesⅣ,Ⅴ,Ⅵ,Ⅶ,Ⅷ,Ⅸ,respectively.The diameters of carbonaceous microspheres used in samplesⅠ,Ⅱ,Ⅲ,Ⅳ,ⅤandⅥare 3μm,and those used in samplesⅦ,Ⅷ,andⅨare 4μm.The temperaturein the fast and medium heating modes is directly increased to 500℃at 2 and 1℃/min respectively,while the temperature in the slow heating mode is increased to 500℃at 1℃/minwith 30 min holding at 400℃.The scale bars are 1μmfor the samples fromⅠtoⅥand 1.3μmfor samples fromⅦtoⅨin the first column.All the scale bars in the second column are 0.5μm,while the scale bars in the third and higher columns are all 0.3μm(a).Illustration of the formation of multishelled ZnOhollow microspheres through different heating processes(b)[57]
模板法可进一步分为软模板法和硬模板法,软模板法的优点是模板成本低,合成条件相对温和。主要缺点是它们的合成基于复杂的溶胶-凝胶过程,并且过渡金属离子的水解和聚合反应难以控制。硬模板法的优点是目标氧化物材料的结构可以通过选择具有所需结构的硬模板来控制。另外由于用作硬模板的介孔二氧化硅高温稳定,从而使许多金属氧化物结晶,因此可以获得具有高度结晶的金属氧化物。目标金属氧化物需要通过NaOH或HF溶液除去二氧化硅模板,尽管可以通过煅烧除去的介孔碳也可以用作硬模板,但介孔碳作为硬模板的主要缺点是前体水溶液对孔壁的润湿性差。此外硬模板法仍然需要引入过渡金属离子前体的固溶步骤,因此将材料范围限制为在溶液中稳定的材料。而且,不能合成与介孔二氧化硅反应的前体材料。对于模板法而言,控制模板的形貌就可以得到相应的纳米材料,因此可以通过多种方法来改变制备过程中的主要影响因素,进而调控纳米粒子的粒径、形貌和结构,从而得到具有目标形貌的金属氧化物。
2.2 无模板法制备不同形貌的金属氧化物
Xiao等[58]以Co(NO3)2·6H2O作为钴源,通过简单地改变NaOH和Co(NO3)2·6H2O的比例而无须使用封端剂,采用一步水热法制备了暴露(001)晶面的立方体,(001)和(111)晶面的截短八面体以及(111)晶面的八面体(图3),根据面心立方纳米晶体的形状主要取决于沿(001)和(111)晶面方向的生长速率之比,对不同形貌的Co3O4的形貌调控机制进行了解释。少量的反应物可能有利于提高沿(111)晶面方向的生长速率并促进暴露(001)晶面的Co3O4立方体的形成,随着Co(NO3)2·6H2O含量的增加,沿(111)晶面方向的生长速率降低,而沿(001)晶面方向的生长速率提高,这导致暴露(001)和(111)晶面的Co3O4截短八面体的形成,当NaOH和Co(NO3)2·6H2O的量再进一步增加时,沿着(001)晶面方向的生长速率显著增加,从而获得暴露(111)晶面的Co3O4八面体。

图3 三种形貌的焙烧后的Co3O4的SEM图像及其结构模型:Co3O4立方体[(a)~(c)],Co3O4截短的八面体[(d)~(f)]和Co3O4八面体[(g)~(i)][58]Fig.3 SEMimagesof the three typesof calcined Co3O4 and their structure models:Co3O4 cubes[(a)~(c)],Co3O4 truncated octahedra[(d)—(f)],and Co3O4 octahedra[(g)—(i)][58]
Liao等[59]通过简便的水热法并在空气中焙烧合成了由许多纳米针构成的新型三维3D中孔Co3O4纳米花,Co3O4多孔纳米针的一端的宽度为120~150 nm,另一端的宽度逐渐变窄,直至呈尖端状。研究了Co3O4微米花随时间增加形貌演变过程:当水热处理1 h,会生成尺寸为50~60 nm的Co3O4纳米颗粒,当反应时间延长至1.5 h,生成长度约为0.6 mm、直径为0.1~0.2 mm的中孔Co3O4纳米棒,有趣的是,继续将时间延长到2 h,就会形成束状Co3O4纳米针,反应3 h后,获得了由纳米针构建的3D分层Co3O4微型花,多孔Co3O4纳米针的密度不够高,因此在纳米花的表面上留下了很多空隙。随着反应时间的进一步增加,纳米花的形貌几乎保持不变,Co3O4纳米针的密度变高。尿素是水溶液中的弱碱,在高温下加热会产生OH-,通过尿素和水之间的水解反应可以有效地控制晶体的生长速率,生成的Co3O4纳米颗粒倾向于聚集以降低其高表面能。这种纳米针的形成可能是由于反应过程中Co2+和OH-的消耗所导致的,详细的机制还需要进一步的研究。Qiao等[60]通过使用钴基层状氢氧化物金属盐(Co-LHMS)作为自模板,制备了纳米棒组装的3D辐射状Co3O4,通过调节前体的水热反应时间获得了片状花形和哑铃状辐射结构的Co3O4。进一步研究从不同的水热反应时间(2~12 h)获得的样品以揭示Co-LHMS的形成机制,从不同时间段获得的Co-LHMS具有三种主要结构,其中Co-LHMS-2h具有均匀的片状花状结构,Co-LHMS-4h具有3D辐射状结构,而CoLHMS-12h具有哑铃形辐射状结构,值得注意的是,Co-LHMS-6h和Co-LHMS-8h都是具有3D和哑铃形辐射状结构的Co-LHMS的混合物。阴离子在反应过程中起重要作用,加热后尿素分解形成氢氧根离子和二氧化碳,然后二氧化碳将与氢氧根离子反应形成碳酸根,最后,二价金属阳离子Co2+将与OH-、CO2-3和H2O结合形成稳定的碳酸钴氢氧化物。当反应时间为2 h,Co2+沿表面的扩散速率(Vdiff)快于沉积位置的沉积速率(Vdep),从而形成片状结构,随着Co2+浓度的降低,更快的Vdep导致晶体的辐射状生长,在4 h形成3D辐射状结构。在生长过程中,CO2-3在控制生长方向中发挥作用,随着反应时间增加,CO2-3变得不足导致生长方向从径向变为轴向,最后反应在12 h达到平衡,几乎所有产物均为哑铃形辐射状Co-LHMS。
Sun等[61]通过水热法随后焙烧制备了具有单壳和双壳纳米结构的中空Fe2O3纳米球。通过控制水热反应时间进一步观察了中空Fe2O3球的结构演变过程:水热反应开始时由K3[Fe(CN)6]水解形成的赤铁矿核易于被NH4H2PO4中的H+离子腐蚀,导致形成无定形核并进一步生长为无定形纳米颗粒,当反应时间为1 h,这些无定形纳米粒子会聚集成球以降低总表面能,随着反应的进行发生Ostwald熟化过程。因为内部颗粒比外部颗粒具有更高的表面能,所以当反应时间为3 h,由纳米颗粒获得了均匀的单壳空心球。当反应时间增加到6 h,由于内表面能量的最小化,单壳空心球中的纳米颗粒进一步熟化,形成了双壳结构。当反应时间延长至12 h,H+蚀刻过程逐渐导致内壳消失,随着反应时间的进一步增加至36 h,再次形成单壳空心球,这是由于酸性环境暴露时间的增加导致的,因此可以通过调节水热反应的时间来控制壳的数量。基于Fe2O3球随水热时间的结构演变过程提出了无定形多壳中空球的形成机理:在一定压力和温度下,[Fe(CN)6]3-解离为Fe3+,随后在酸性水溶液中水解为非晶态的Fe2O3纳米颗粒,Ostwald熟化过程驱动了从单壳到双壳中空结构的初始结构演变。随水热时间的增加H+的蚀刻作用超过了熟化过程,这导致了这些中空结构的反向重复出现。Cao等[62]通过水热法制备FeOOH前体,再进一步通过焙烧制备3D分层多孔α-Fe2O3纳米片(图4)。通过分析提出其结构和几何形貌的一个演变过程:首先,Fe2+被部分氧化成Fe3+,这导致在空气环境下搅拌过程中进一步形成Fe(OH)3。将铜板浸入制备的溶液中时,形成的Fe(OH)3可能会黏附在铜板上,并在成核过程中充当晶种的来源,残余的Fe2+与OH-(由尿素水解产生)和O2反应,形成FeOOH,同时Fe(OH)3在水热过程中转化为FeOOH。由于水解形成的初级粒子是热力学不稳定的,因此相邻的初级纳米粒子首先聚集成立方体状的纳米粒子,并进一步长成花状十二边形,在表面能最小化的驱动下,花状十二边形由于Qstwald熟化机制而生长并分裂,随反应时间进一步增加,大块花瓣分裂成层状纳米片,在它们之间留有足够的空间以形成3D框架。由于焙烧后释放H2O,纳米片变得多孔,独特的多孔3D分层纳米结构改善了电子-离子传输,减轻了电极在循环时的体积变化引起的内部机械应力,并在循环过程中形成了3D导电网络,将其作为锂离子电池的负极材料显示出高可逆容量、优异的倍率性能和长期可循环性。
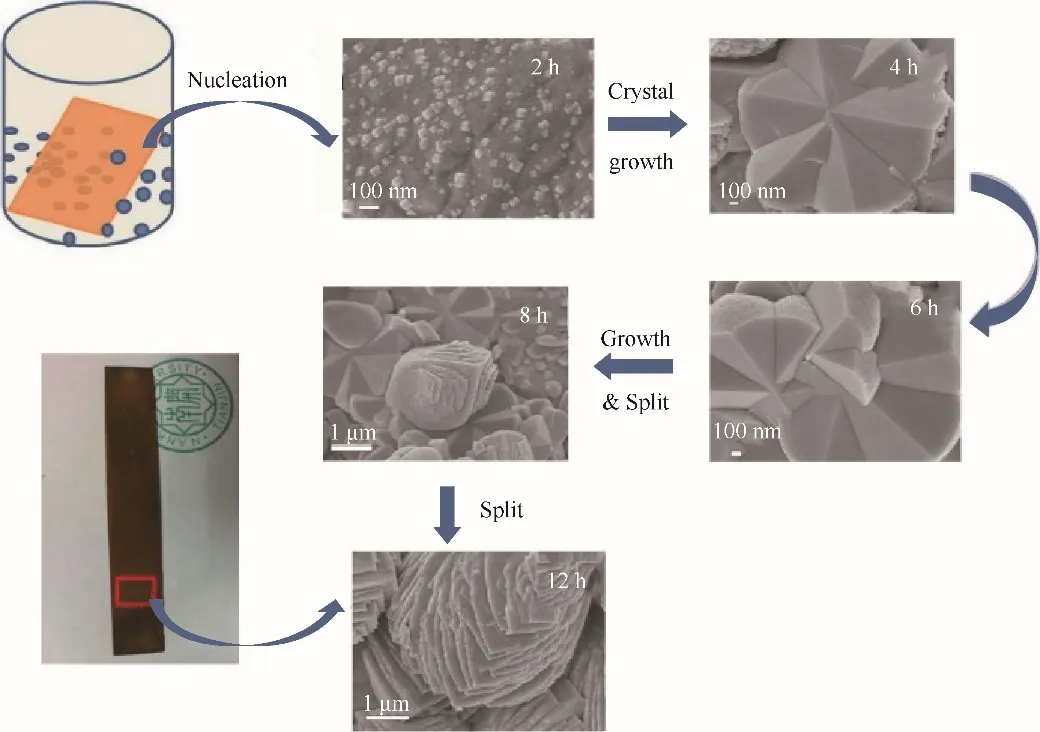
图4 形成前体FeOOH的示意图[62]Fig.4 Schematic illustration of the formation of the precursor FeOOH[62]
Zhao等[63]通过没有任何模板的简单水热法制备了一种新型的3D空心聚集球形WO3纳米片。增加HNO3浓度使得产物WO3(H2O)0.33转变为单斜WO3,直到反应完成,同时颗粒的形貌和尺寸从球形变为纳米片,最终聚集为3D中空球形结构。通过增加H+浓度来抑制(010)优势晶面生长,因此WO3·H2O倾向于以片状结构存在,在随后的干燥过程中,WO3·H2O因缺乏结晶水,形成单斜WO3片。Wang等[64]通过溶剂热法制备了具有双蛋黄结构的ZnO空心球(图5),由于在反应体系中不包含模板和表面活性剂,因此特殊结构的形成归因于气泡辅助的Ostwald熟化机理。在系统能量最小化的驱动下,小的ZnO纳米晶体聚集在一起形成3D微球,初始的非致密聚集体转化为两个较小的封装聚集体,进行该过程的详细原因需要进一步研究,在形成较大的实心球之后,Ostwald熟化成为形成这些空心结构的主要驱动力,这种力可以归因于初始聚集体内部存在固有密度变化。当气体流量变弱时,可能会削弱堆积密度的影响以及聚集体中ZnO纳米晶体的分布,形成具有单蛋黄结构的ZnO空心球,与双蛋黄结构的ZnO空心球相似,单蛋黄结构的ZnO空心球的结构形成分为三个阶段:(1)ZnO纳米晶体;(2)包含较小实心球的非致密ZnO聚集体;(3)具有单蛋黄结构的空心球。这与高压釜中产生的气泡量显著降低是一致的,此外,当高压釜中没有气体排出时,只形成单壳ZnO空心球。

图5 ZnO纳米晶体随时间演变为具有双蛋黄结构的ZnO空心球:1 h(a),12 h(b),24 h(c),相应演变过程的示意图(d)[64]Fig.5 Time-dependent evolution of ZnOnanocrystals to ZnOhollow sphereswith double-yolk egg structure:1 h(a),12 h(b),24 h(c).The corresponding schematic graphs of the evolution process(d)[64]
Liao等[65]通过使用正丁醇钛(TBT)和乙酸(HAc)的简单酸热方法成功制备了由纳米棒和纳米颗粒组成的分层锐钛矿型TiO2球,克服了纳米颗粒的动力学和光散射限制以及一维纳米结构的表面积限制。TBT和HAc之间的反应将首先形成六配位的络合物Ti6O6(Ac)6(OBu)6(R1),随后产物H2O可以进一步与Ti6O6(Ac)6(OBu)6的配合物反应生成Ti6O6(Ac)6(OBu)6-n(OH)n(0≤n≤6)(R2),随后的缩合反应形成纳米带。进行了与时间有关的实验,以观察分层的TiO2球的形成过程,经过3 h的酸热反应后,获得了纳米颗粒,随着反应时间的延长(6 h),形成了纳米带,最终导致了分层球体(12 h)。基于SEM、透射电子显微镜(TEM)、热重-红外(TG-IR)和FTIR分析由纳米棒和纳米颗粒组成的TiO2分层球的可能形成过程,在HAc存在下,TBT初始水解时会形成许多Ti复合中间体的纳米微晶,随着反应时间的进一步延长这些纳米微晶将通过定向附着过程形成纳米带,由纳米带组成的Ti复合中间体分层球将逐渐形成,最后通过基于纳米带的分层Ti-复合中间球的热处理工艺(3 h)获得了由锐钛矿型TiO2纳米棒和纳米颗粒组成的分层TiO2球。Ziarati等[66]通过包括共沉淀制备赤铁矿纳米立方体的多步方法合成了黑色空心纳米立方TiO2(BHC-TiO2)结构(图6)。合成过程包括以下主要步骤:(1)共沉淀法制备Fe2O3纳米立方体;(2)用TiO2动力学控制包覆Fe2O3以生成Fe2O3/TiO2核/壳纳米立方体;(3)HCl水热蚀刻工艺以产生空心二氧化钛立方体;(4)氢处理以获得BHC-TiO2纳米结构。

图6 制备BHC-TiO2的总流程图[(a)~(d)]:相应的BHC-TiO2制备程序的FESEM[(e)~(h)],BSE-SEM[(i)~(l)]和TEM[(m)~(p)]图。白色箭头表示破裂的空心立方结构。(e)~(h)比例尺为1μm,(i)~(p)比例尺为500 nm[66]Fig.6 Overall flowchart for fabrication of BHC-TiO2[(a)—(d)];Corresponding FESEM[(e)—(h)],BSE-SEM[(i)—(l)]and TEMimages[(m)—(p)]of the BHC-TiO2 fabrication procedure.The white arrows indicate the cracked hollow cubic structures.The scale bars are 1μm[(e)—(h)]and 500 nm[(i)—(p)][66]
无模板法调控形貌主要通过加入添加剂作为形貌指向剂来控制形貌。添加剂种类包括无机阴离子、表面活性剂、螯合剂等或通过控制晶体生长的条件(如前体的种类、反应体系、反应温度、反应时间、反应物浓度、溶剂等)来调控材料形貌。与模板法相比,无模板法不需要模板的去除,实验过程更加简单,但在无模板法中产物形貌受诸多因素影响,因此合成特定形貌的金属氧化物较为困难。
2.3 物理法制备不同形貌的金属氧化物
Guo等[67]通过微波辅助的金属有机框架(MOF)方法控制MIL-53(Fe)形貌演变,微波辐照提供了均匀的晶核,并通过MOF模板提供了有限的孔隙率和骨架,通过简单地调整微波辐射时间即可获得各种形貌的多孔Fe2O3纳米结构,包括纺锤体、凹面八面体、固体八面体、核壳八面体和纳米棒(图7)。微波辐射时间对于MIL53(Fe)前体的形成起着重要作用,对MOF衍生的铁氧化物的形貌也具有重要作用,MIL-53(Fe)的结构中每个Fe离子由来自四个H2BDC配体的四个氧原子和两个μ2桥连的氧原子进行六配位,而两个相邻的Fe离子则通过两个螯合的羧基和一个μ2-O桥连接,因此,沿a轴形成一维Fe-O之字形链,这进一步在BDC配体的连接下构建了三维网络。因此,MIL-53(Fe)倾向于表现出纺锤形或八面体相关的形貌,通过分析提出核壳八面体形貌的Fe2O3-2和Fe2O3-6的形成机理:核壳结构的形成主要归因于煅烧过程中的两个相反的力,即黏附力和收缩力,这导致外面的壳和里面的核分离,形成Fe2O3-2核壳多孔八面体,黏附力和收缩力作用在MIL-53(Fe)-6的核壳八面体中不明显,导致从MOF前体继承多孔的核壳八面体结构的Fe2O3-6形成。

图7 MIL-53(Fe)的生长过程及其相应的铁氧化物的形貌(a)以及Fe2O3-2和Fe2O3-6的形成过程的示意图(b)[67]Fig.7 Schematic illustration of the process of MIL-53(Fe)growth and morphology of their corresponding iron oxides(a)and the formation process of Fe2O3-2 and Fe2O3-6(b)[67]
Liu等[68]通过脉冲激光沉积制备不同形貌的ZnO,通过逐级改变脉冲激光的输入能级以及对材料状态的观察确定成核和晶体生长过程中的不同能垒,通过精确调节克服特定能垒的脉冲激光能量,可以在无催化剂的环境中在基底上获得具有所需取向和形貌的ZnO晶体。晶体在激光诱导的生长中表现出不同的行为,并且生长速率比其他水热反应高几个数量级,通过分析反应/扩散受限区域中的晶体生长特性,他们定义了一组准则来调整ZnO晶体的尺寸(图8)。当输入功率低于9.55 kW/cm2时,所有晶体表面都以非常低的速率被活化,并且晶体将均匀生长并形成球形结构。当功率从9.55 kW/cm2增加到15.92 kW/cm2时,克服了棱柱面(m面)生长的能垒,从而导致了从圆柱体到六边形形貌的转变。当脉冲激光功率增加到22.29 kW/cm2时,晶体将沿着轴的优先生长方向生长,克服了(0001)面生长的能垒,使(0001)面的生长速度急剧增加。当脉冲激光功率增加到22.29 kW/cm2以上时,尖端不再平滑,而是受到由Miller指数较高的面组成的小丘的束缚,并且晶体尖端趋向于(101ˉ1),小丘面的比表面自由能比(0001)面低,因此这些较高的Miller指数面占优势。当脉冲激光功率高于28.66 kW/cm2时,(0001)表面具有最高的生长速率,从而导致形成金字塔状的晶体结构。值得注意的是,克服势垒的是峰值功率而不是累积的能量,即使总输入能量相同,在特定的高功率密度下生长的某些形貌在较长的时间里积累的较低功率的照射条件下不会出现。晶体生长通常被认为是岛的2D成核过程,然后是台阶的横向运动,岛的2D成核过程类似于初始的非均质成核过程,其中自由能垒与特定平面的表面能相关,棱柱面(101ˉ0)的表面能为1.15 J/m2,(101ˉ1)平面为1.37 J/m2,基面(0001)为2.0 J/m2,因此,用于平面的晶体生长的能垒顺序为(0001)>(101ˉ1)>(101ˉ0)。实验结果表明,随着能量输入的增加,晶面的出现遵循(101ˉ0),(101ˉ1),(0001)的顺序,与上面引用的表面能值顺序相同。通过调节激光能量以选择性克服不同表面的能垒,可以获得不同形貌的晶体,克服动力学障碍后,晶体将保持热力学稳定状态,从而使总表面自由能最小化,通过根据反应和扩散受限区域中的不同机制调整激光功率密度,可以获得具有所需尺寸和形貌的ZnO。
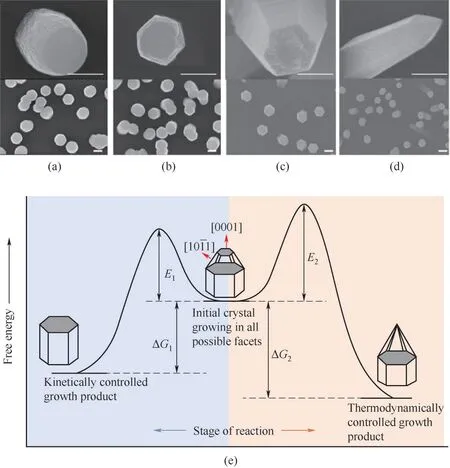
图8 在9.55 kW/cm2(a),15.92 kW/cm2(b),22.29 kW/cm2(c),28.66 kW/cm2(d)和200 kHz的激光功率密度下生长5 min的ZnO晶体的SEM图像。动力学控制(左侧)和热力学控制(右侧)过程中晶体生长的示意图(e)。所有比例尺代表长度均为500 nm[68]Fig.8 SEMpictures of ZnOcrystals grown under laser power density of 9.55 kW/cm2(a),15.92 kW/cm2(b),22.29 kW/cm2(c),28.66 kW/cm2(d)in 200 kHz for 5 min.Schematic illustration of crystal growth in kinetically controlled(left direction)and thermodynamically controlled(right direction)processes(e).All scale bars represent 500 nm in length[68]
Chen等[69]通过直接液相注入化学气相沉积法制备Co3O4薄膜,并探究了气相沉积温度对薄膜晶体结构和形貌的影响。与其他物理方法相比,直接液相注入化学气相沉积(DLI-CVD)是大规模制备和具有较高台阶覆盖度的膜的更经济的技术,不仅可以精确地控制化学成分和沉积速率,而且还大大降低了对反应气氛和底物的要求(图9)。(220)、(311)、(111)、(400)和(511)的TC(哈里斯纹理系数TC用于计算取向度)值波动,并且在Tdep=723 K和823~973 K时,在TC=1(随机取向)周围显示出相对不规则的分布。与之相对,在Tdep=773 K时,TC(111)的最高值为2.1,即完美取向的42%,这与所有其他取向的TC值不同,从而验证了在773 K下制备的Co3O4膜沿(111)优先生长。此外在Tdep=773 K下制备的(111)取向的Co3O4薄膜的表面微观结构与其他具有随机取向的Co3O4薄膜明显不同,所以提出了一个假设,即具有三角形形状的独特纳米壁结构必须来源于成核过程并且可以由有效的(111)平面外取向确定。放大的场发射扫描电子显微镜(FESEM)图像为(111)取向的Co3O4晶粒形貌演变提供了线索,可以清楚地观察到分散的三角形面,并发现它们变为纳米壁结构。解释了Co3O4薄膜的(111)取向生长机理:立方结构中对于(111)方向,有8个可能的矢量方向,它们将体心与原始立方单元中的8个角原子相连,当尖晶石型Co3O4沿(111)方向生长时,垂直于极轴的(111)平面可以穿过晶胞并形成(100)平面的三角形[图9(b)红色三角形单元],另一侧基于(100)和(111)晶面具有相反指向的“三角形金字塔”沿(111)方向生长[图9(b)绿色三角形单元]。通常,这两个金字塔构成了沿(111)方向进一步形成晶粒的基本单元,这两个基本的金字塔单元不仅可以彼此耦合生长,而且还可以填充晶粒之间的不匹配以补偿残余应力。因此在外延(111)取向的立方氧化物膜中,经常选择单晶基底用于薄膜的外延生长,以减少自成核步骤的残余应力并调节生长取向。然而在这项研究中选择非晶态熔融石英来研究Tdep与Co3O4薄膜的生长行为之间的内在联系,在这种情况下,临界成核温度(Tcn)点对于Co3O4薄膜的定向生长至关重要,因为不存在来自非晶石英衬底的影响。与外延生长不同,在773 K的Tcn处Co3O4晶粒可以沿(111)面外取向自由生长,而没有高度有序的面内取向,因此可以看到三角形的微观结构。如果成对的三角形(111)晶粒中的一些晶粒尺寸不对称地生长,则可以基于这两个三角形单元形成纳米墙构筑单元,并以不确定的面内图案展开。
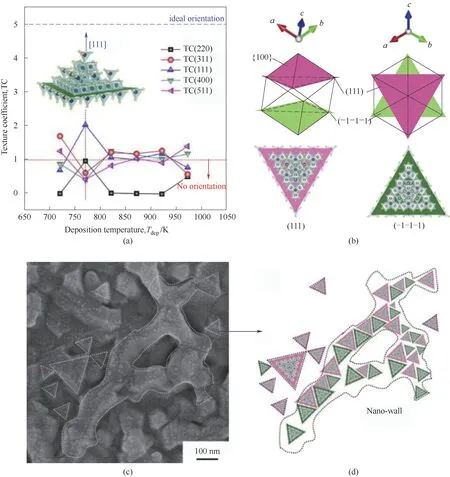
图9 T dep对Co3O4薄膜的TC(220)、(311)、(111)、(400)和(511)的影响(a),(111)面的生长机理(b),放大的FESEM图像(c)和在773 K下制备的Co3O4薄膜的纳米墙结构示意图(d)[69]Fig.9 Effect of T dep on TC(220),(311),(111),(400)and(511)of Co3O4 films(a),growth mechanismof(111)planes(b),enlarged FESEMimage(c)and schematic of nanowall structure of Co3O4 film prepared at 773 K(d)[69]
Li等[70]以CoCl2·6H2O为前体,通过简便的一步式超声喷雾热解工艺合成了具有不同初级粒径的纳米粒子组装的Co3O4微球(在600、750、850和950℃下获得的产物分别表示为S-600,S-750,S-850和S-950)(图10)。此外研究发现粉末收集器的温度显著影响产品中的氯含量,氯化物残留的形成机理有两种可能的解释:(1)氯化钴不完全分解。(2)在镍泡沫过滤器中发生的逆反应,因为在反应器出口处,来自废气的气态HCl对Co3O4的氯化在热力学上是有利的,Co3O4粉末的多孔结构还有利于气态HCl和蒸汽的吸附。所有样品均由平均大小约为2 mm的次级球形颗粒组成,这些颗粒由许多初级纳米颗粒组装而成,反应器温度对次级颗粒尺寸没有明显影响。与之相对,随着温度从600℃增加到950℃,初级粒径逐渐增加,S-600、S-750、S-850和S-950的初级粒径分别约为20、80、120和180 nm,比表面积从14.30 m2/g减少至1.81 m2/g。通过对比微球中心或边缘确定所有样品都具有多孔结构,结构的内部空心程度随着反应器温度的升高而增加,S-600具有填充的内部结构,而S-750、S-850和S-950具有空心结构。因此通过改变合成温度是一种简便的一步法控制粉末的初级粒径和内部结构的方法。初级粒径对初始库仑效率、比容量和速率性能有重要影响,具有最小粒径和低Cl含量的Co3O4在200 mA/g的电流密度下经过50次循环后显示出最高的初始库仑效率73%和1340 mA·h/g的高可逆容量。

图10 在不同温度下制备的样品的SEM和TEM图像:S-600[(a),(b)],S-750[(c),(d)],S-850[(e),(f)]和S-950[(g),(h)][70]Fig.10 SEM and TEM images of samples prepared at different temperatures:S-600[(a),(b)],S-750[(c),(d)],S-850[(e),(f)]and S-950[(g),(h)][70]
Yang等[71]通过两步电沉积方法制备了分层氧化锌纳米棒阵列(ZNRAs),用作柔性染料敏化太阳能电池(DSSC)光电阳极。通过沉积初级ZNRAs,然后在初级ZnO纳米棒表面上生长次级分支的ZnO纳米棒,实现了两步电沉积。控制不同的电沉积持续时间制备的初级和分级ZNRAs的SEM图像表明随着沉积时间从900 s增加到3600 s,初始ZnO纳米棒的平均直径和长度增加,但密度降低,尺寸的增加归因于较长的时间积累,而密度的降低归因于ZnO纳米结构通过Zn(OH)2-xx的脱水最终沉积在工作电极上的逆反应使较小的纳米棒溶解而导致的。所有分层的ZNRAs都表现出花状的分层ZnO纳米棒结构,而次级纳米棒的尺寸则随着初级ZNRAs沉积时间的增加而增加,涂覆纳米晶种层后,具有较大密度和较小尺寸的初级ZnO纳米棒具有较大的表面积,形成更多的ZnO纳米颗粒,并且这些纳米颗粒会以ZnO晶核的形式在二次电沉积过程中生长。然而具有紧凑分布的一级结构不利于次级纳米棒的轴向生长,并且次级电沉积溶液中Zn2+的总量是确定的,结果导致形成短而细的针状次级纳米棒。与之相对,尽管密度较小但尺寸较大的初始ZnO纳米棒的纳米晶种层涂层表面积较小,但稀疏分布为次级ZnO纳米棒的轴向和径向生长提供了更大的空间,导致更长的沉积时间和更粗的次级纳米棒的形成。
物理法制备避免了引入杂质的过程,而且原料利用率较高,得到的产物均一性较好。但是一方面其适用范围较窄,难以制备具有特定形貌的材料,另一方面物理法需要的原料和仪器比较昂贵,能耗较大,这些缺点阻碍了其发展与应用。
3 不同形貌的金属氧化物的应用
3.1 在加氢反应中的应用
加氢催化剂在生物质资源的高效转化及综合利用和二氧化碳加氢制高附加值碳氢化合物中具有重要作用。Fei等[72]以立方介孔二氧化硅KIT-6作为模板,通过纳米浇筑法合成了一系列有序介孔氧化钼(MoO3)复合物,用于环己烯加氢反应(所得的介孔MoO3材料表示为Mo-m,其中Mo表示介孔MoO3,m表示相应的硬模板KIT-6的水热温度)。6 h后,样品Mo-100、Mo-120、Mo-80和MoO3的转化率分别达到100%、92%、71%和28%,转化率随MoO3催化剂的表面积和孔体积的增加而增加,因此认为介孔MoO3的催化氢化性能优于块状MoO3归因于更高的比表面积和3D相连的介孔结构,该结构为与环己烯和氢分子相互作用提供了更多的活性位点。Nguyen-Huy等[73]以KIT-6和SBA-15作为模板,通过纳米浇筑法合成了介孔钴氧化物[由SBA-15和KIT-6模板制备的催化剂样品分别称为m-Co3O4(p6mm)和m-Co3O4(Ia3d),在350或500℃的温度下还原1 h,并将获得的产物标记为m-CoO(p6mm)-350,m-CoO(Ia3d)-350,m-Co(p6mm)-500和m-Co(Ia3d)-500(根据晶体结构,空间群和还原温度]。X射线吸收近边结构光谱(XANES)表明m-Co3O4(p6mm)和m-Co3O4(Ia3d)在350℃时会部分还原,而在500℃时会完全还原。XRD、TEM、XPS、H2-TPR,X射线吸收光谱(XAS)和原位X射线衍射(in situXRD)表明m-CoO(p6mm)-350主要为CoO相,而m-Co(p6mm)-500包含金属Co和在表面上的一小部分CoOx。与之相反,由于在350℃时的显著还原,m-CoO(Ia3d)-350具有金属Co但主要为CoO相。对于被完全还原m-Co(Ia3d)-500,几乎没有CoOx。m-CoO(p6mm)-350的转化率(41%)比m-CoO(Ia3d)-350(33.6%)高,尽管m-CoO(p6mm)-350的表面积较小,但其质量活性是m-Co3O4(p6mm)的21倍,比m-CoO(Ia3d)-350高1.6倍。还原温度进一步提高到500℃导致活性降低,这归因于表面积的降低以及CoO相的减少,结果表明氧化钴还原性显著影响介孔结构,对于糠醛(FAL)加氢活性,CoO物种比表面积更重要。DFT计算证实了FAL在CoO(111)比在Co(111)的吸附能力强,不仅导致FAL转化率高,而且对2-甲基呋喃(MF)的选择性也发生明显变化。由于与Co(111)相比,糠醇(FA)在CoO(111)上的C—OH键容易裂解,CoO位点可诱导FAL选择性加氢为FA并随后产生MF。Meijboom等[74]通过逆表面活性剂胶束法合成了介孔锰氧化物、钴氧化物和锰钴复合氧化物,用于使用叔丁基过氧化氢作为氧化剂的苯乙烯选择性氧化反应中。Mn-Co复合氧化物的H2-TPR峰归因于Mn4+和Co3+的还原,介孔氧化锰和介孔氧化钴的峰位移归因于通过库仑和配位相互作用的锰和钴之间的电子相互作用。催化活性以Mn-Co_350(66.6 h-1)>MnO2_350(47.4 h-1)>Co3O4_350(35.6 h-1)的顺序降低,使用介孔锰钴氧化物获得的高催化活性归因于两种金属产生的协同作用。热处理温度对氧化锰催化剂对氧化苯乙烯产物的选择性的影响可以忽略。对于介孔氧化钴催化剂,热处理温度对介孔钴氧化物催化剂对苯乙烯氧化物产物的选择性影响很大,因为与在550℃下煅烧的催化剂相比,在低温下煅烧的催化剂对苯乙烯氧化物的选择性更低且随着介孔氧化钴的煅烧温度升高。热处理温度对介孔复合氧化物催化剂对苯乙烯氧化物的产物选择性可忽略。认为随着热处理温度升高,对苯乙烯氧化物的选择性增加可归因于氧化钴的电子结构的变化,介孔Mn-Co氧化物对苯乙烯或苯甲醛的选择性降低则归因于金属与金属之间的相互作用导致电子结构的变化。
Zhang等[75]制备了三种具有不同载体形貌的Pt/CeO2催化剂并将其应用到硝基苯的液相加氢反应中。通过电镜表征确定CeO2纳米棒的优势暴露晶面为(110)和(100),CeO2纳米立方体的优势暴露晶面为(100)以及纳米颗粒上优势暴露晶面为(111)和(100),不同的暴露晶面具有不同的还原性,根据前人的报道,不同CeO2晶面上阴离子空位的形成能遵循(110)<(100)<(111)的顺序,这意味着氧空位更容易在高能晶面上形成,高能晶面很容易被还原以产生更多的Ce3+表面位点和氧空位,由于更强的金属-载体相互作用,不仅有利于Pt物种的分散和稳定,而且还为反应物和中间体提供了更多的吸附位点。提出了Pt/CeO2催化的硝基苯加氢途径,在该途径中硝基苯直接加氢成N-苯基羟胺,然后进行歧化和缩合步骤形成乙氧基苯,最后通过乙氧基苯加氢分解成苯胺。通过H2-TPR研究发现CeO2的还原性表面作为电子给体,Ce3+的较高电子密度或氧空位促进了硝基苯和N-苯基羟胺的吸附,而由于N-苯基羟胺比硝基苯具有更高的电子密度,N-苯基羟胺在还原的Ce表面上的吸附更困难。通过改变催化剂预处理温度,高温还原的远离Pt的Ce3+位点可以有效地吸附N-苯基羟胺,而那些低温还原与Pt相邻的Ce3+位点相对较弱地吸附N-苯基羟胺(图11)。结合硝基苯和N-苯基羟胺吸附的漫反射傅里叶变换红外光谱(DRIFTS),具有较高电子密度的N-苯基羟胺只能强烈吸附在具有较强电子给予能力的Ce3+表面上,高温还原会在远离Pt的位置产生额外的Ce3+位点,从而可以有效吸附N-苯基羟胺并促进苯胺的形成。使得在600℃还原的Pt/CeO2-R-600纳米棒催化剂在室温下硝基苯加氢反应中显示出优异的苯胺产率[40.8 mol/(g·h)]和出色的稳定性。

图11 高温还原产生的额外的Ce3+位点有利于反应物和中间体的吸附的示意图[75]Fig.11 Additional Ce3+sites derived from high-temperature reduction favoring adsorption of the reactant and intermediate[75]
Li等[76]以CeO2纳米棒和纳米立方体为载体,通过浸渍法制备了两种不同形貌的3.0%(质量)Pd/CeO2负载型催化剂,用于环境压力下100~200℃的温度范围内气相苯加氢反应。CeO2纳米棒主要暴露(110)和(100)晶面,而CeO2纳米立方体主要暴露(100)晶面,暴露的不同晶面会导致不同的氧空位浓度以及纳米CeO2载体表面上金属Pd的存在形式,最终影响金属Pd与纳米CeO2载体之间相互作用的强度。H2-TPD证实了3.0%(质量)Pd/CeO2催化剂具有优异的氢吸附-脱附性能,可以为加氢反应提供更多的活性氢物种。3.0%(质量)Pd/CeO2纳米棒和纳米立方体催化剂在200℃时保持100%的环己烷选择性条件下,苯加氢转化率分别达到96.4%和91.2%,相应的反应速率分别为4.15×10-7mol/(g·s)和3.92×10-7mol/(g·s)。Ouyang等[77]以Co3O4纳米棒和纳米片为载体,通过浸渍法制备了两种不同形貌Pt/Co3O4催化剂,用于2 MPa和190~230℃下CO2加氢制备高级醇反应。Co3O4纳米片主要暴露晶面为(112),Co3O4纳米棒的主要暴露晶面为(011)。H2-TPR和XPS证实了载体形貌显著影响了其氧化还原行为,Co3O4纳米片比Co3O4纳米棒更容易被还原。Pt/Co3O4纳米棒催化剂的CO2转化率高于Pt/Co3O4纳米片催化剂,但Pt/Co3O4纳米棒的醇选择性较低。根据前人的报道,金属Co催化CO2加氢主要产物是甲烷,因为它具有较强的加氢能力和RWGS活性,因此为了提高醇的选择性,将反应温度降低至200℃,以抑制金属Co的形成。Pt/Co3O4纳米片催化剂在200℃时获得最高的高级醇时空产率[0.56 mmol/(g·s)],其中甲醇的存在比例最大(78%~93%),其次是乙醇,丙醇和丁醇的比例非常低,对于这两种催化剂,较高的醇(R2+OH)选择性随着反应温度的降低而降低。
Liu等[78]制备了四种结构明确的不同形貌和暴露晶面的CeO2载体(CeO2纳米棒、立方体、八面体和多 面 体,分 别 表 示 为CeO2-R、CeO2-C、CeO2-O、CeO2-P),通过浸渍法制备了四种不同形貌的Pd/CeO2催化剂(图12),用于CO2还原制备甲醇的反应,并深入探究了载体形貌对Pd存在形式和氧空位的影响。CeO2-R的优势暴露晶面为(110)和(111)晶面,而CeO2-P的优势暴露晶面为(111)和(100)面,CeO2-C和CeO2-O的优势暴露晶面分别为(100)和(111)晶面。考虑到氧空位对于CO2活化起重要作用,结合XPS、拉曼(Raman)和储氧量(OSC)表征证实了氧空位的形成过程及其对晶面的依赖性,其中CeO2纳米棒具有最低的氧空位形成能,并且具有最高密度的表面氧空位数量。DFT表明与Pd1/CeO2和PdxCe1-xOδ固溶体相比,在实际反应条件下金属Pd纳米颗粒是最稳定的物种。Pd的存在极大促进了氧空位的形成,但是它不能改变氧空位浓度的顺序,即2Pd/CeO2-R>2Pd/CeO2-P>2Pd/CeO2-C>2Pd/CeO2-O,并且(111)和(100)晶面的界面处的氧迁移率高于单个(111)和(100)面。CO2加氢反应是Pd表面上H2的吸附/活化与CeO2载体氧空位上CO2的吸附/活化之间的协同反应,催化活性主要取决于二氧化铈表面氧空位上CO2的活化,氧空位的形成能越低,则氧空位的浓度越高,但是一个氧空位的反应性越低。因此,合适的形成能有利于CO2活化,尽管在2Pd/CeO2-R催化剂上氧空位的反应性较低,但由于具有大量的氧空位该催化剂仍显示出最高的催化活性,在2Pd/CeO2-R上,当气体体积空速为6 L/(g·h)时,可获得最高的甲醇时空产率为22.8 mg/(g·h)。

图12 CeO2-R[(a),(b)],CeO2-P[(c),(d)],CeO2-C[(e),(f)]的TEM和高分辨率TEM(HRTEM)图像,CeO2-O的SEM(g)和HRTEM(h)图像,插图为CeO2-R,CeO2-P,CeO2-C和CeO2-O的暴露晶面[78]Fig.12 TEMand high-resolution TEM(HRTEM)images of CeO2-R[(a),(b)],CeO2-P[(c),(d)],CeO2-C[(e),(f)],SEM(g)and HRTEM(h)images of CeO2-O.The insets schematically illustrate the crystal planes exposed on the CeO2-R,CeO2-P,CeO2-Cand CeO2-O[78]
Chai等[79]制备了两种不同形貌的ZnO(纳米片和纳米针),通过浸渍法获得具有相似金粒径和不同金/金属氧化物界面的两种催化剂(Au/ZnO-P和Au/ZnO-N),以探究ZnO晶面对乙炔加氢活性的影响。高分辨透射电子显微镜(HRTEM)表明ZnO-P中极性面为主要暴露晶面,而ZnO-N中非极性面是主要暴露晶面。电子顺磁共振(EPR)结果表明金在ZnON上的沉积可能导致Vo•形成顺磁的Vo0,这可能会促进金属金在非极性面上的稳定化。原位漫反射傅里叶变换红外光谱表明在Au/ZnO-N上CO在金属金上的吸附强度比在Au/ZnO-P上强,因此针状ZnO中非极性平面稳定金属金的能力可能是Au/ZnO-N催化性能更优异的原因。Jia等[80]制备具有暴露晶面分别为(101)、(100)和(001)面的锐钛矿型TiO2纳米晶体的Ir/TiO2催化剂,用于巴豆醛的选择性加氢反应。NH3-TPD表明催化剂表面酸性位点的数量顺序依次为Ir/TiO2-(100)>Ir/TiO2-(101)>Ir/TiO2-(001),此外与TiO2载体相比,Ir/TiO2的NH3-TPD曲线差别较大,证实了Ir/TiO2催化剂的表面酸度受TiO2载体暴露的晶面影响。具有较高表面酸度的Ir/TiO2-(100)(0.55 mmol/g)比具有较低表面酸度的Ir/TiO2-(101)(0.51 mmol/g)的活性低,可能是反应物或产物分子在催化剂表面上的强烈吸附和/或脱羰反应引起的CO中毒引起的活性位点的占据所致。前人研究表明氧空位倾向于接受电子,因此充当Lewis酸位点,这种由氧空位产生的Lewis酸位点通常伴随着邻近晶格氧产生的表面Lewis碱位点。EPR表明反映三种Ir/TiO2催化剂中表面氧空位的相对浓度的色心浓度F1+顺序依次为Ir/TiO2-(100)>Ir/TiO2-(101)>Ir/TiO2-(001),证实了Ir/TiO2催化剂的表面酸度与催化剂中的氧空位浓度一致。巴豆醛(CRAL)吸附的原位FTIR光谱表明CRAL在Ir/TiO2催化剂的吸附强度与TiO2晶面有很强的相关性。另一方面,Ir/TiO2催化剂上的CRAL脱附曲线与相应的TiO2载体上CRAL类似的脱附曲线可以解释这种相似性。此外当引入H2时,CRAL吸附在Ir/TiO2催化剂上1658 cm-1处的谱带迅速降低,因此作者认为Ir-TiOx界面位点在反应中,特别是在提高巴豆醇(CROL)选择性方面起重要作用。
3.2 在氧化反应中的应用
一氧化碳(CO)氧化因其在基础研究和实际应用中的重要意义和价值,成为多相催化领域中研究最多的反应之一。Liu等[81]通过简便的水热法合成了具有不同粒径不同形貌的赤铁矿(α-Fe2O3)纳米立方体和纳米棒,合成的不同粒径的α-Fe2O3纳米棒和纳米立方体分别称为LC(大立方体)、SC(小立方体)、LR(大纳米棒)和SR(小纳米棒)。α-Fe2O3-LC是被六个与(012)面等效的晶体面包围的平行六面体,α-Fe2O3-SC被六个等效晶体面[(110)和两个等效(001)面]包围,尽管α-Fe2O3-LR和α-Fe2O3-SR具有相似的暴露晶面(110),但是α-Fe2O3-SR的(110)面比例比α-Fe2O3-LR低。α-Fe2O3-LR显示出比其他样品更优异的CO催化氧化性能。Fe原子密度相对较高的特定暴露晶面对催化性能的影响更大,由于(110)晶面上有高密度铁原子,(110)晶面的比例高表明更多的活性氧种类和更高的催化活性。May等[82]制备了优势暴露晶面为(100)面的CeO2纳米立方体(1CuCe NC)和为(110)面的CeO2纳米棒(1CuCe NR),以探究CeO2晶面对CuO-CeO2催化CO氧化活性的影响。CO-TPD证实了1CuCe NC催化剂上的CO2吸附比1CuCe NR催化剂上的CO2吸附更容易。此外与1CuCe NR催化剂相比,1CuCe NC催化剂在低温下形成二氧化碳的峰值强度要强得多。前人研究表明形成的CO2量与催化剂表面上存在的氧气浓度直接相关,因此1CuCe NC催化剂的表面氧含量明显高于1CuCe NR催化剂的表面氧含量,这些丰富的表面氧物种促进了CO2的形成,并且与1CuCe NR催化剂相比,CO2更容易解吸有助于1CuCe NC催化剂的优异的催化性能,证实了CO的吸附显著取决于CeO2纳米结构的晶面,并且在优势暴露晶面为(100)面的1CuCe NC上是有利的。利用XPS和in situDRIFTS表征证实CO分子在还原的Cu(Ⅰ)位上的吸附是影响表面氧结合的关键因素,因此主要暴露晶面为(100)面的1CuCe NC催化剂比主要暴露晶面为(110)面的1CuCe NR催化剂对CO的氧化活性高得多。Zheng等[83]合成了具有不同粒径、氧空位浓度、氧迁移率和优势暴露晶面的三维有序大孔(3DOM)CeO2和CeO2-ZrO2复合氧化物(这些合成后的CeO2和CeO2-ZrO2样品分别表示为C450和CZ450,将C450和CZ450样品分别在600和800℃的空气中分别 煅 烧2 h,分 别 表 示 为C600、C800、CZ600和CZ800)作为模型催化剂,以探究CeO2基催化剂上CO氧化的结构依赖性和反应机理。TEM分析表明C450、C600、CZ450和CZ600样品的(110)/(110+111)之比分别为23%、20%、8%和45%,XPS证实了Ce3+/(Ce+Zr)以及Oads/Olat的比例按CZ600>C450>C600>CZ450的顺序降低,这与催化剂的活性顺序完全一致,这表明Ce3+浓度以及晶格氧浓度对于确定催化性能起着非常重要的作用。通过连续CO脉冲模式进行了储氧量(OSC)测量,总储氧量按CZ600>C450>C600>CZ450的顺序降低,证实了优势暴露晶面和氧空位浓度是决定催化活性的关键因素。Raman和XPS证实了CZ600具有丰富的氧空位,可以吸附O2分子。CO-TPR检测到CO和表面OH基团之间的表面水煤气变换反应,CO-TPR过程中in situDRIFTS的结果表明存在甲酸盐类物种,甲酸盐类物种被认为是从CO到碳酸盐的中间体。尽管不能忽略Mars-van Krevelen机理,但Langmuir-Hinshelwood机理应该是3DOM CeO2基催化剂上CO氧化的关键反应途径。根据L-H机理,CO分子首先吸附在催化剂表面上,然后与吸附的氧反应,最后将形成的CO2解吸到气相中。
特定氧化物和复合氧化物均具有优异的CO氧化活性,但其高温稳定性较差,且易被水和硫化物毒化,严重限制了其实际应用。而负载型贵金属基催化剂具有较高的CO氧化活性。Wang等[84]通过水热法可控地合成了具有不同形貌的Co3O4(立方体、花状、片状和矩形分别表示为Co3O4-C、Co3O4-F、Co3O4-P、Co3O4-R),通过浸渍法制备了四种不同形貌的Pd/Co3O4催化剂,用于CH4和CO氧化反应。较高的CO氧化活性与催化剂优异的氧化还原能力有关,H2-TPR证实了Pd/Co3O4-R的氧化还原能力最强,XPS证实了Pd/Co3O4表面Oads/Olat摩尔比顺序为Pd/Co3O4-C>Pd/Co3O4-R>Pd/Co3O4-P>Pd/Co3O4-F。对于CO氧化反应,与相应的空白载体相比Pd/Co3O4-R、Pd/Co3O4-F、Pd/Co3O4-P催化活性显著下降,而Pd/Co3O4-C催化活性明显提升。而在甲烷氧化过程中,Pd的加入增加了Co3O4的活性,而浸渍前后的活性顺序基本保持一致,并且Co3O4到Pd/Co3O4的活性提高程度与表面Oads/Olat的顺序一致,表明Pd和具有不同形貌的Co3O4发生的相互作用不同。根据前人的研究催化剂表面的Oads/Olat比以及暴露的晶面在甲烷燃烧中起着至关重要的作用,可变氧化态的金属阳离子通过提供表面晶格氧阴离子并从气相中抽出氧气来同时还原可用于氧化还原反应的氧化物离子,不同的晶面具有不同的原子排列,因此对于不同的晶面,分子氧的活化和表面或晶格氧阴离子的类型存在差异。通常,表面晶格氧是高温下烃氧化的关键物质,参与选择性氧化,而吸附的亲电物质O22-、O2-和O-等负责低温下的烃氧化,并优先参与整个氧化过程。Gao等[85]制备了纳米球(S)和纳米棒(R)两种不同形貌的α-Fe2O3,通过浸渍法制备了两种不同形貌的Au/α‐Fe2O3催化剂。吡啶吸附的红外光谱证实了α‐Fe2O3(R)具有更强的Lewis酸性并且α‐Fe2O3(R)的Lewis酸位点密度是α‐Fe2O3(S)的两倍左右,较高表面Lewis酸位点密度可以使金稳定在α‐Fe2O3(R)上,α‐Fe2O3(R)的表面比α‐Fe2O3(S)的表面更不平整,催化活性与α‐Fe2O3(R)表面缺陷位点的存在有关,具有较低的氧结合能的缺陷位点可能与催化CO氧化循环中CO2形成有关,由于较强的金属-载体相互作用,Au/α‐Fe2O3(R)比Au/α‐Fe2O3(S)包含更小的半球形金纳米颗粒,Au/α‐Fe2O3(R)不仅在CO氧化中更具活性,而且在反应过程中也表现出了更高的稳定性。Au/α‐Fe2O3(R)在25℃下已经具有活性(转化率为12%),在125℃下的CO转化率为98%。与之相对,Au/α‐Fe2O3(S)的活性增加较低,在125℃时CO转化率仅为17%。
Ha等[86]通过水热法可控地合成了两种不同形貌的CeO2(立方体和八面体),通过浸渍法制备了两种不同形貌的Au/CeO2催化剂,用于CO氧化反应。在200~300℃的温度范围内,Au/CeO2立方体的TOF高于Au/CeO2八面体。在300℃时,Au/CeO2立方体的TOF为0.69 s-1,是Au/CeO2八面体(0.17 s-1)的四倍。DFT计算证实了Au-CeO2界面介导的CO氧化通过MvK机制活化(图13)。H2-TPR表明Au/CeO2立方体优异的催化活性归因于Au-CeO2(100)界面增强的氧气释放能力,这有助于通过MvK机制进行CO氧化。根据实验分析(H2-TPR和XPS)以及Au和CeO2之间的电子相互作用及其对氧空位形成能(Evac)的影响的理论解释,Au和CeO2之间的电子相互作用可以改变Au/CeO2催化剂的Evac。基于DFT的微动力学建模和实验,在不同的反应条件[p(CO)和反应温度]下构建TOF/速率图证实了CeO2(100)晶面在Au-CeO2界面活化CO氧化优异的协助能力,尽管由于CO中毒CO更倾向于吸附在Au NPs,但在Au/CeO2立方体和Au/CeO2八面体中都观察到p(CO)与CO氧化的实验TOF值之间存在正相关,此外用于CO氧化的氧气由Au-CeO2界面提供,并且Au-CeO2界面处增加的CO浓度可增强Au/CeO2催化剂的CO氧化活性。
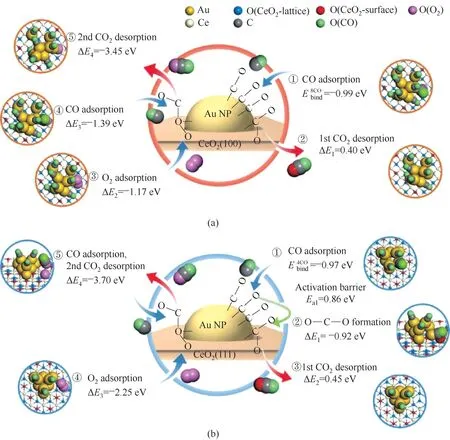
图13 具有8或4个吸附的CO分子的Au/CeO2(100)(a)和Au/CeO2(111)(b)催化的示意性CO氧化途径表明尽管Au/CeO2(111)对于附加的O—C—O形成步骤是必须的[(b)图,步骤②],但CO氧化发生在Au-CeO2界面[86]Fig.13 Schematic COoxidation pathways catalyzed by Au/CeO2(100)(a)and Au/CeO2(111)(b)with 8 or 4 adsorbed COmolecules show that COoxidation occursat the Au-CeO2 interface,although an additional O—C—Oformation step[(b),step②]is required forAu/CeO2(111)[86]
Gu等[87]制备了三种具有规则形貌的α-Fe2O3(截短的六边形双锥、准立方体和六边形片,分别表示为α-Fe2O3-THB、α-Fe2O3-QC、α-Fe2O3-HS)(图14),随后通过沉淀-沉积法制备了三种不同形貌的Au/α-Fe2O3催化剂,用于CO氧化反应。基于SEM图像估算的边缘长度,对于α-Fe2O3-THB,整个表面的七分之三属于(113)面,而表面的七分之二和七分之一分别属于(214)和(104)面。对于α-Fe2O3-QC,表面的每三分之一分别属于(104),(110)和(0120)面。对于α-Fe2O3-HS,整个表面的九分之七属于(001)晶面,而表面的九分之二属于(110)晶面。在体积空速(GHSV)为30000 ml/(g·h),100℃时,Au/α-Fe2O3-THB、Au/α-Fe2O3-QC和Au/α-Fe2O3-HS上CO2生成速率分别为0.77、0.08、0.27 mmol/(g·s)。通过O2/表面羟基-TPD、CO-TPSR和in situFT-IR证实了表面氧/羟基物质在不同界面上形成关键中间体的作用,不同的中间体(CO2-3和HCO-2)的变化直接受界面特征的控制,即弱吸附的氧和表面羟基物质以及特定的Au-Fe2O3边界结构,从而决定了CO的活化并转化为二氧化碳。DFT计算表明水吸附在载体上比在Au上更有利,因此表面羟基的性质与载体的表面结构或Au-载体边界结构密切相关。结合O2-TPD证实了在α-Fe2O3-THB和α-Fe2O3-HS载体以及Au/α-Fe2O3-THB和Au/α-Fe2O3-HS的界面上有反应性羟基,认为这种类型的羟基在(113)和(001)面上在能量上是有利的,因为这些平面的羟基化会降低其表面能。DFT计算也进一步证实了α-Fe2O3-THB的(113)面的表面原子密度最低,因此暴露出高度开放的表面结构,Au沉积后,O—O距离在Au-载体边界处扩展,所有这些结构特征都有利于分子氧的吸附/表面羟基化,以最大限度地减少系统的表面能,Au上活化的CO容易与边界和/或相邻羟基周围的氧空位上的弱吸附氧反应,生成Ⅱ型碳酸盐(CO2-3)以及甲酸盐物种,也说明了Au/α-Fe2O3-THB在CO氧化方面优于另外两个催化剂。

图14 α-Fe2O3-THB样品的TEM图[(ⅰ),(ⅱ)],HRTEM图(ⅲ),IFFT图像(ⅳ)(a).α-Fe2O3-QC样品的TEM图[(ⅰ),(ⅱ)],HRTEM图(ⅲ),IFFT图(ⅳ)(b).α-Fe2O3-HS样品的TEM图(ⅰ),SAED模式(ⅱ)和示意图(ⅲ)(c)[87]Fig.14 TEM images[(ⅰ),(ⅱ)],HRTEMimage(ⅲ),IFFTimage(ⅳ)of theα-Fe2O3-THBsample(a).TEM images[(ⅰ),(ⅱ)],HRTEM image(ⅲ),IFFT image(ⅳ)ofα-Fe2O3-QCsample(b).TEM image(ⅰ),SAEDpattern(ⅱ),and schematic illustration(ⅲ)ofα-Fe2O3-HSsample(c)[87]
金属氧化催化剂的暴露晶面和晶体缺陷可能会影响催化剂的性能。在大多数情况下,氧迁移率是影响氧化过程的关键因素。金属氧化物的氧化活性通常与氧空位有关的Mn+/M(n+1)+离子对的存在有关,氧空位对于反应物分子的吸附以及异裂以及O2的活化至关重要,而可还原性金属氧化物在本质上具有两种金属价态,即使在没有贵金属的情况下也可以催化氧化反应进行。活性位点的密度取决于晶体的表面,因此氧化物的形貌在氧化反应中起主要作用。实现可控的金属-金属和金属-氧相互作用的金属氧化物以用于新的应用是目前的研究热点及难点。
3.3 在蒸汽重整反应中的应用
Zhang等[88]通过使用PVP辅助的溶剂热法制备了暴露(10-10)非极性面的针状ZnO微晶(ZnO-N),以无任何极性面的市售ZnO(ZnO-P)作为对照,随后通过浸渍法制备了一系列具有不同Pd负载的Pd/ZnO催化剂。ZnO-N的优势暴露晶面为(100)、(002)和(101)面,这表明颗粒沿(0001)方向生长,使极性ZnO表面位于针状ZnO微晶的尖端。商用ZnO的晶面为(100)、(002)和(101)面,这表明实质上ZnO-P没有优势暴露的极性或非极性面,ZnO-N样品的非极性面比市售ZnO-P的比例高,使用具有不同比例非极性晶面的ZnO-N和ZnO-P两种稳定载体研究ZnO晶面对PdZn合金形成的影响及其在MSR中的催化性能(图15)。对于Pd/ZnO-P和Pd/ZnO-N催化剂,随着Pd负载量的增加,CO选择性降低,因为较高的Pd负载量通常会导致PdZn的粒径更大,这在MSR中表现出较低的CO选择性。在相同的Pd负载量下Pd/ZnO-N催化剂比Pd/ZnO-P具有更高的CO选择性。更重要的是,即使与具有相似粒径的PdZn相比,Pd/ZnO-N的CO选择性也比Pd/ZnO-P的CO选择性高。在具有相似PdxZny粒径的2%(质量)Pd/ZnO-N和1%(质量)Pd/ZnO-P催化剂上,CO选择性均随甲醇转化率的提高而降低,在所研究的甲醇转化率范围内,在相同的甲醇转化率下,Pd/ZnO-N催化剂上的CO选择性远高于Pd/ZnO-P催化剂,表明ZnO晶面在MSR的反应路径中也起着关键作用。结合CO-FTIR,CO氧化反应和HRTEM表征证实了通过改变Pd的负载量,在ZnO的非极性(10-10)面上会形成各种组分的PdxZny相,在低Pd负载[<4.8%(质量)]时,富Pd相(PdxZny,x>y)占优势,x/y比随Pd负载的增加而降低,导致Pd/ZnO-N催化剂的CO选择性降低。在高Pd负载[例如9.1%(质量)Pd/ZnO-N]下,尽管可以形成PdZnβ相,但其他富含Pd的PdxZny甚至是分离的Pd共存,导致高的CO选择性,在Pd/ZnO-N催化剂上观察到高的CO选择性是由于形成了不同组成的富Pd相导致的。另一方面,发现在极性(0001)面上容易形成稳定的PdZnβ,随着Pd含量的增加,Pd会沉积在极性(0001)面上,从而在ZnO-P载体上形成稳定的PdZnβ,因此需要较高的Pd负载量[即4.8%(质量)Pd/ZnO-P]以形成稳定的PdZnβ相,该相促进了CO2的形成并因此降低了Pd/ZnO-P催化剂上的CO选择性。
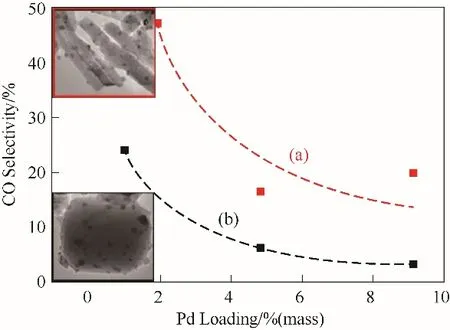
图15 在甲醇转化率为30%~40%时,Pd/ZnO-N(a)和Pd/ZnO-P(b)的CO选择性(反应条件:100~200 mg催化剂,甲醇浓度6.4%(mol),催化剂质量/进料气流速=0.037~0.123 g·s/ml,T=250℃)[88]Fig.15 COselectivity of Pd/ZnO-N(a)and Pd/ZnO-P(b)with Pd loading amount under methanol conversion of 30%—40%[88]
Kourtelesis等[89]通过水热法制备了不同的二氧化铈纳米结构(CeO2纳米立方体、CeO2纳米棒和CeO2纳米花,分别表示为CeO2-NC、CeO2-NR和CeO2-FL),并通过沉淀法获得了CeO2(表示为CeO2-PPT),随后通过浸渍法制备一系列具有不同载体形貌的Pt/CeO2催化剂用于低温乙醇蒸汽重整。结合瞬时质谱(Transient-MS)和FT-IR光谱揭示乙醇蒸汽重整的反应机理,不同形貌的CeO2不影响乙醇蒸汽重整反应网络,CeO2载体催化乙醇脱氢和乙醇脱水导致乙烯形成,然后发生分解和重整反应。通常与CeO2-PPT相比,纳米结构的载体具有更高的活性,反应开始时所需要的温度略低。程序升温表面反应(TPSR)表明不同形貌的Pt/CeO2上反应路径基本相同,但是载体形貌会影响催化活性,对于Pt/CeO2-NC乙醇脱氢的起始温度为430 K,对于Pt/CeO2-NR为370 K,对于Pt/CeO2-FL催化剂为460 K,对于Pt/CeO2-PPT为300 K,这表明脱氢活性遵循PPT>NR>NC>FL的顺序。此外高温CH4峰与CO甲烷化反应的催化剂活性相关,并且与在相同温度区域的第二个CO峰相关,CH4峰的高强度和CO峰的低强度表明催化剂对CO甲烷化具有很高的活性,这些峰受载体形貌影响很大,对于NC、NR和FL催化剂,第二个CO峰的强度较低,而Pt/CeO2-PPT催化剂在该区域显示出高浓度的CO,表明Pt/CeO2-NC,Pt/CeO2-NR和Pt/CeO2-FL具有很高的CO甲烷化反应速率,其次是Pt/CeO2-PPT,因此Pt/CeO2-PPT催化剂对乙醛分解和CO甲烷化的活性最低。四种不同形貌的Pt/CeO2催化剂用于低温乙醇蒸汽重整反应机理是相同的,FTIR表明首先发生乙氧基物质的分解及其氧化形成乙酸盐的过程,随后乙酸盐物质的分解和/或其氧化为碳酸盐物质的分解可能进一步分解为CO2,但是在空白二氧化铈载体上所形成的乙酸盐不会分解或转化为其他物种。所有催化剂对乙醇的低温乙醇蒸汽重整反应均具有活性,但催化活性随载体形貌的变化而变化。Pt/CeO2-NC表现出最高的初始乙醇转化率,而Pt/CeO2-PPT是活性较低的催化剂,对于所有催化剂,均观察到H2、CH4、CO、CO2、乙醛和痕量丙酮,但是产物的分布与载体的形貌相关,H2、CO和CH4是所有催化剂形成的主要产物,表明乙醇分解或乙醇脱氢生成乙醛,然后分解为CH4和CO是在573 K下发生的主要反应,而在Pt/CeO2-PPT上观察到明显的乙醛形成表明该催化剂促进了乙醇脱氢。
Wang等[90]通过水热法制备了CeO2纳米棒(表示为CeO2-NP),并通过沉淀法获得了CeO2纳米颗粒(表示为CeO2-NR),随后通过浸渍法制备了两种具有不同载体形貌的Ir/CeO2催化剂用于乙醇蒸汽重整。晶面分析表明CeO2纳米颗粒的优势暴露晶面为(111),CeO2纳米棒的优势暴露晶面为(110)和(100),H2O-TPD表明对于正向反应是OH基团向金属纳米颗粒的溢流,而逆向反应是金属纳米颗粒辅助的二氧化铈羟基化的可逆步骤,与在CeO2纳米颗粒上解吸的水相比,在CeO2纳米棒上解吸的水更多,这是因为纳米颗粒上(100)面的暴露相对较少,从而产生较少的氧空位活化了较少的水分子。水基本上吸附在二氧化铈载体表面上,需要金属-载体界面以提供表面氧与乙醇中间体反应,因此Ir/CeO2纳米颗粒催化剂比Ir/CeO2纳米棒催化剂具有更高的水活化能力,对于乙醇重整反应表现出更优异的催化性能。Ir/CeO2纳米棒催化剂在673 K下形成乙醛和氢,主要发生乙醇脱氢,在此温度下没有产生C1产物,表明与纳米棒催化剂相比,纳米棒催化剂上C—C键断裂能力相对较弱。当温度升至773 K时,产生氢气、甲烷、二氧化碳和丙酮,表明乙醛裂解为C1产物并冷凝为丙酮,CO的浓度远低于CH4的浓度,表明在此温度下发生了水煤气变换反应。从773~873 K,乙醛和丙酮几乎被转化,CH4浓度的少量降低表明发生了甲烷蒸汽重整反应,而CO2的明显增加表明发生了水煤气变换反应。873 K以上时,CO浓度持续升高,而CO2浓度降低,表明发生了逆水煤气变换反应。在923 K下产物分布为53%H2、14%CO、18%CO2和6%CH4。催化活性以及反应路径与用于乙醇蒸汽重整反应的Ir/CeO2催化剂的结构(主要来源于二氧化铈载体的形貌)密切相关,在Ir/CeO2纳米颗粒催化剂上获得的乙醇转化率和氢浓度比在纳米棒催化剂上获得的乙醇转化率和氢浓度更高表明Ir/CeO2纳米颗粒催化性能更优异。稳定性测试表明CeO2纳米棒维持了初始的形貌,而CeO2纳米棒变成了多面体表明CeO2纳米棒在蒸汽重整条件下的结构稳定性相对较差。
蒸汽重整反应是一个复杂的反应体系,目前用于该反应的催化剂主要是负载型金属基催化剂,但在反应条件下易发生活性组分烧结、积炭而导致失活。利用钙钛矿型氧化物、混合金属氧化物、尖晶石型氧化物等新型催化剂的多金属协同作用可以有效地减少积炭生成,因此非负载型氧化物催化剂的开发引起了研究者的广泛关注。
Zhao等[91]制备了双钙钛矿型氧化物La1.6Sr0.4FeCoO6作为载氧体用于化学循环甲烷蒸汽重整(CL-SMR)联产合成气和氢气。结合XRD、XPS、H2-TPR以及TG表征手段证实了用Sr替代La可以诱导B和B'位形成具有高价态的金属,例如Fe5+、Fe4+和Co3+,这些位点为甲烷的解离提供了活性位点,同时双钙钛矿结构中Fe-Co多金属的共存诱导大量氧空位而协同促进了氧的扩散,La1.6Sr0.4FeCoO6的还原性、氧气迁移率和蒸汽分解反应性得到了有效提高。在固定床反应器中进行等温反应分析反应产物确定了三个反应阶段,包括甲烷被活性吸附氧完全氧化、甲烷被晶格氧部分氧化以及甲烷分解。对于CL-SMR反应中的载体提供活性氧,一种可能的反应途径是甲烷与活性吸附氧的完全氧化产生CO2和H2O,甲烷与晶格氧的部分氧化产生CO和H2,甲烷分解形成碳沉积。在还原阶段,晶格中的氧从主体转移到边界,参与甲烷还原反应,同时其中的一部分转变为性质更稳定的化学吸附氧。甲烷的解离与晶格氧的扩散同时伴随大量合成气的产生,有效地提高了催化剂的抗积炭能力。足够的活性位点可以破坏蒸汽的H—O键。氧空位可以由H2O的O原子立即提供。同时由H—O键断裂产生的两个H原子结合在一起形成H2。
金属氧化物可以促进甘油的蒸汽重整,并且随着金属氧化物表面碱度的提高气态产物的转化率增加。Padmakar等[92]采用共沉淀法通过改变MgOSrO的摩尔比并保持Co3O4的含量恒定来制备一系列Co-Mg-Sr混合金属氧化物催化剂(催化剂分别表示为CMS331、CMS321、CMS311和CMS312,其中C、M和S表示Co、Mg和Sr,而数字表示其在催化剂中的摩尔比),用于甘油蒸汽重整(GSR)产氢。TPR表明与其他催化剂相比,CMS311催化剂中存在大量高度分散和相互作用的氧化钴位点。通过CO2-TPD证实了催化剂的碱度顺序为CMS311>CMS312>CMS321>CMS331,MgO和SrO摩尔比相等的催化剂显示出最高的碱度,并且随着样品中MgO含量的增加,碱度会降低。CMS311催化剂具有最佳的活性,实现了100%的甘油转化率和72%的氢产率。在700℃下,CMS311催化剂的H2和CO2比已达到理论值,因此认为催化剂的高活性归因于较强的金属-载体相互作用,粒径较小的钴颗粒的存在和碱性,MgO在增加钴金属分散性方面起重要作用,SrO在控制金属与载体的相互作用、减少积炭形成和增加碱度方面起着至关重要的作用。
Xi等[93]制备含有少量CuO相的Cu-Al尖晶石型氧化物(新鲜催化剂命名为“CAx-T”,其中x和T分别表示Al/Cu原子比和煅烧温度)已成功地用于甲醇蒸汽重整(MSR)。通过H2-TPR证实了Cu-Al尖晶石催化剂中少量CuO相在低温下引发甲醇转化,CuAl2O4逐渐生成活性Cu。CO2-TPD表明还原的CA2.0-900对CO2的较强吸附主要是由于CuAl2O4中释放出Cu后,多孔的Al2O3所致。MSR中Cu-Al尖晶石催化剂对CO选择性也随之相应地变化。在MSR反应过程中,CA2.5-900的活性和副产物CO的生成速率都是先升高然后降低,而CA4.0-900和CA6.0-900则一直降低,此外CA2.5-900的催化性能和可再生性优于CZA(Cu-Zn-Al催化剂)。Yang等[94]通过使用溶胶-凝胶法合成了一系列不同化学计量比的Mn-Cr-O尖晶石型催化剂,用于乙烯的蒸汽重整反应。结合XRD、XPS和XANES表征证实了在化学计量的MnCr2O4中引入额外的Mn导致Mn1.5Cr1.5O4的尖晶石晶格中的Mn3+被取代,而在Mn0.5Cr2.5O4中的过量Cr形成Cr2O3。动力学研究表明在Mn-Cr-O尖晶石催化剂上在大气压和873 K下进行的乙烯蒸汽重整在乙烯中是一级,在水中为负级,而在过量的氢气中为零级。反应动力学与Mars-van Krevelen机理一致。尖晶石催化剂的性能优于纯金属氧化物,因为在相同的重整条件下Cr2O3几乎是惰性的,而Mn3O4则由于原位还原为MnO而失活。他们提出了双位点Mars-van Krevelen型机理,其中H2O对蒸汽重整活性的轻微抑制作用归因于H2O在尖晶石表面的吸附比C2H4更强。与MnCr2O4和Mn1.5Cr1.5O4相比,Mn0.5Cr2.5O4中过量的Cr2O3导致其在乙烯蒸汽重整反应中失活,并显著提高了活化势垒。在类似的蒸汽重整条件下,乙烷在Mn0.5Cr2.5O4催化剂上脱氢成乙烯,而对蒸汽重整没有明显的反应性。在Mn1.5Cr1.5O4上观察到尖晶石型催化重整的最高TOF值归因于Mn3+的存在。尽管Mn0.5Cr2.5O4样品中存在Cr2O3,但Mn0.5Cr2.5O4和MnCr2O4相似的催化性能表明化学计量的尖晶石是两种催化剂表面的活性位。
目前研究新型的蒸汽重整反应,由于蒸汽重整反应过程中活性中心烧结和催化剂失活,维持催化剂稳定性是面临的最大难点。对于负载型金属氧化物催化剂,提高载体与催化剂之间的相互作用可以有效地减少活性中心的烧结。与之相比,非负载型金属氧化物催化剂包括钙钛矿型氧化物、混合金属氧化物、尖晶石型氧化物等新型催化剂的多金属协同作用可以有效地减少积炭,稳定性较高。
4 结 论
金属氧化物催化剂因其特性在工业催化领域展现出巨大的优势和广阔的应用前景,引起了国内外研究者的广泛关注。形貌依赖的金属氧化物为微调催化活性位提供了一种新策略,优先暴露出反应晶面的形貌可控的金属氧化物缩小了模型催化剂与实际催化剂之间的差距,有利于阐明内在的构效关系。基于形貌可控的金属氧化物特性,可以获得对目标反应具有高活性和高选择性的金属氧化物催化剂。本文综述了不同形貌的金属氧化物的制备方法、生长机制及其结构特性,重点研究其溶剂热合成过程,总结了可能的形成机制,深入理解不同形貌的金属氧化物形成规律,进而能在理论上指导特定形貌材料的可控合成。聚焦于金属氧化物催化剂在氧化反应、加氢反应和蒸汽重整反应中的最新研究进展,为高性能金属氧化物催化剂的设计和合成提供了一定的依据。
尽管调控金属氧化物催化剂的形貌可在许多重要的催化反应中获得较为理想的催化效果,但仍然存在一些迫切需要解决的问题和挑战。(1)在机械层面上很难追踪晶体生长过程动态演变,然而深入理解纳米材料的成核和生长过程对于调控反应体系通过相、组成和形貌控制制备特定形貌纳米结构氧化物是必要的。(2)反应物种类与添加量、添加剂的种类与添加量、反应时间、反应温度等众多因素都会影响金属氧化物的最终形貌,虽然研究者对于金属氧化物的生长机制及其结构特性有一定认识,但是很难达成统一的理论和规律性认识,严重制约了特定形貌的金属氧化物载体形貌的精确调控。(3)由于多相催化反应过程中催化剂的表面结构一般会随着其所处的环境变化而变化,永远处于一种动态平衡中,即表面重构(包括吸附诱导重构、前处理重构、反应环境重构等),使得在反应条件下对于表面活性位点的表征和确定变得十分困难。(4)大多数特定形貌的金属氧化物催化剂尚处于实验室研究阶段,距真正工业化应用还有很长一段距离,不同形貌的金属氧化物催化材料的工业应用仍然是今后研究的重点和难点。
近年来,用于机械研究的原位方法库也不断扩大,通过原位衍射技术可以准确高效地实时监测氧化物形成过程。基于这些问题和挑战,研究的重点可从以下几个方面着手。(1)可采用原位XAS和小角X射线散射/广角X射线散射(SAXS/WAXS)技术提供有关晶体成核生长过程的关键信息,并将其应用于氧化物材料的形貌控制的制备瓶颈中。通过越来越多的实例阐明反应参数对所得氧化物材料的相和形貌的影响,得出一般合成策略对于形貌控制的趋势。(2)采用VASP、Materials Studio Modeling等软件通过模拟晶体的内部结构来预测晶体材料的外部晶形,可用于研究外部因素对晶体生长的影响,通过进行晶面控制预测,进而为可控合成提供指导。(3)通常制备的金属氧化物纳米材料并不是完美晶体,其暴露的晶面并不完美,会产生各种各样的缺陷,如台阶、空位、晶格扭曲、弯曲、多晶等。这些缺陷的存在通常与其独特的催化性能具有很大的关联。此外金属氧化物在不同化学反应环境中、不同催化反应阶段均会发生表面重构现象,即表面重构是一种持续的动态平衡的过程。因此,有必要采用有效的原位技术手段对金属氧化物表面结构进行表征,近常压扫描隧道显微镜(STM)和环境透射电镜技术(ETEM)研究表面重构过程,原位时间分辨漫反射红外光谱和原位/操作条件XAFS等手段可获得实际反应条件下活性中心的结构演化及表面缺陷类型以及配位结构变化等信息。(4)为推进不同形貌的金属氧化物催化剂的工业化应用,合成稳定性、效率和可靠性是实现氧化物纳米材料大规模工业生产的重要标准,当务之急是开展系统的研究,以获得调控合成参数的指导原则,实现金属氧化物催化剂的表面积和表面结构控制,制备在催化条件下具有高稳定性的不同形貌的氧化物结构,以促进金属氧化物催化材料在催化领域的应用。
猜你喜欢
陶瓷学报(2021年4期)2021-10-14
粉末冶金技术(2021年3期)2021-07-28
陶瓷学报(2021年1期)2021-04-13
粉末冶金技术(2021年1期)2021-03-29
陶瓷学报(2020年6期)2021-01-26
物理实验(2019年7期)2019-08-06
World Journal of Diabetes(2019年7期)2019-07-23
航空材料学报(2019年2期)2019-04-15
中学化学(2015年9期)2016-04-14
云南民族大学学报(自然科学版)(2015年4期)2015-11-14
