
1-(2-甲基-4-苯基-1H-吡咯-3-基)乙酮的合成
2021-06-29 01:54邓护军马利娟王军绪田真真
化学与生物工程 2021年6期
邓护军,马利娟,王军绪,田真真,刘 斌
(1.西安万隆制药股份有限公司,陕西 西安 710119;2.陕西国际商贸学院医药学院,陕西 西安 712046)
吡咯及其衍生物是一类非常重要的五元含氮杂环化合物。作为一种常见的分子母核结构,多取代吡咯结构广泛存在于各种药物分子中,例如降血脂药物阿托伐他汀(Atorvastatin)[1-2]、抗肿瘤药物他莫司汀(Tallimustine)[3-4]、非甾体类抗炎药物托麦汀(Tolmetin)等[5](图1)。多取代吡咯结构也被大量地应用于药物分子的设计合成,如钾离子竞争性酸阻断剂[6]、HIV-1整合酶抑制剂[7]、α7烟碱型乙酰胆碱受体调节剂[8]、抗肿瘤活性分子[9-10]等,均取得了很好的效果,研究者因此对多取代吡咯类化合物的合成和应用研究更加重视[11-12]。

图1 含有多取代吡咯结构的药物
吡咯类化合物的常用合成方法有Paal-Knorr吡咯合成[13]、Hantzsch吡咯合成[14]、Barton-Zard吡咯合成[15]等,这些方法均具有一定的底物局限性。为了合成结构新颖的多取代吡咯类化合物、研究其在药物分子设计合成中的应用,作者以β-硝基苯乙烯(Ⅱ)和乙酰丙酮为原料,在有机碱条件下,通过Michael加成反应得到中间体Ⅲ,再经过硝基还原、关环反应得到目标化合物1-(2-甲基-4-苯基-1H-吡咯-3-基)乙酮(Ⅰ),中间体及目标化合物结构经1HNMR和MS表征,并着重考察碱催化的β-硝基苯乙烯和乙酰丙酮的Michael加成反应条件。合成路线见图2。

图2 1-(2-甲基-4-苯基-1H-吡咯-3-基)乙酮的合成路线
1 实验
1.1 试剂与仪器
β-硝基苯乙烯、乙酰丙酮、1,8-二氮杂二环十一碳-7-烯(DBU)、N,N-二异丙基乙胺(DIEA)、三乙烯二胺(DABCO)、三乙胺(TEA),泰坦科技股份有限公司;铁粉(60~80目),无锡亚秦联合化工有限公司;柱层析硅胶(300~400目),青岛海洋化工厂;其它试剂均为市售分析纯。
AV300型核磁共振仪(氘代溶剂为CDCl3),德国布鲁克公司;Ultima Global Spectrometer型质谱仪(ESI源),美国沃特斯公司;RE-52AA型旋转蒸发仪,上海亚荣生化仪器厂;SHB-Ⅲ型循环水式多用真空泵,郑州长城科工贸有限公司。
1.2 合成方法
1.2.1 3-(2-硝基-1-苯基乙基)戊烷-2,4-二酮(Ⅲ)的合成
将2.98 g(20.0 mmol)β-硝基苯乙烯、2.20 g(24.0 mmol)乙酰丙酮加入到25 mL二氯甲烷(DCM)中,搅拌均匀后加入4.04 g(40.0 mmol)TEA,在25 ℃下反应3 h,薄层层析法(TLC)监测反应结束;将反应体系减压浓缩,所得粗品通过硅胶柱层析(V石油醚∶V乙酸乙酯= 5∶1)分离纯化,得到4.78 g淡黄色固体化合物Ⅲ,收率96.2%,熔点130~132 ℃。1HNMR(400 MHz,CDCl3),δ:7.35~7.29(m,3H),7.19(d,J=6.8 Hz,2H),4.68~4.61(m,2H),4.39~4.37(m,1H),4.28~4.22(m,1H),2.30(s,3H),1.94(s,3H);ESI-MS,m/z:250.33[M+H]+。
1.2.2 1-(2-甲基-4-苯基-1H-吡咯-3-基)乙酮(Ⅰ)的合成
将2.49 g(10.0 mmol)中间体Ⅲ溶于15 mL乙醇中,加入2.80 g(50.0 mmol)铁粉,搅拌下加入5 mL冰醋酸,然后升温至60 ℃后搅拌4 h,TLC监测反应结束;将反应体系倒入20 mL冰水中,然后加入碳酸氢钠水溶液调节至碱性,减压抽滤,滤液用乙酸乙酯(3×30 mL)萃取,减压浓缩,所得粗品经硅胶柱层析(V石油醚∶V乙酸乙酯=4∶1)分离纯化,得到0.96 g淡黄色固体化合物Ⅰ,收率48.1%。1HNMR (400 MHz,CDCl3),δ:11.01(br,1H),7.33~7.27(m,3H),7.22~7.20(m,2H),6.62(s,1H),2.37(s,3H),1.93(s,3H);ESI-MS,m/z:200.31[M+H]+。
2 结果与讨论
2.1 碱的种类和用量对中间体Ⅲ收率的影响
碱的种类和用量对Michael加成反应合成中间体Ⅲ的影响较大。由于两种反应底物活性较高,所以选择碱性较低的有机碱(DBU、DIEA、DABCO和TEA)进行Michael加成反应,考察其用量对中间体Ⅲ收率的影响,结果见表1。

表1 碱的种类和用量对中间体Ⅲ收率的影响
由表1可知:(1)4种有机碱均能较好地促进Michael加成反应,中间体Ⅲ收率较高(≥81.0%),其中,使用DABCO和TEA得到的收率接近,考虑到后处理难易程度(TEA沸点低于90 ℃,通过减压旋蒸即可除去;而DABCO沸点高于170 ℃,通常需通过柱层析纯化),选择TEA为反应用碱。(2)当TEA用量n(TEA)∶n(β-硝基苯乙烯)为2.0∶1时,收率为96.2%;增加TEA用量n(TEA)∶n(β-硝基苯乙烯)为2.2∶1时,收率略有降低;但减少TEA用量n(TEA)∶n(β-硝基苯乙烯)为1.8∶1时,收率明显降低。因此,确定适宜的TEA用量n(TEA)∶n(β-硝基苯乙烯)为2.0∶1。
2.2 物料比对中间体Ⅲ收率的影响
在n(TEA)∶n(β-硝基苯乙烯)为2.0∶1、25 ℃反应3 h的条件下,考察物料比[n(乙酰丙酮)∶n(β-硝基苯乙烯)]对中间体Ⅲ收率的影响,考虑原料成本,选择乙酰丙酮过量,结果见表2。
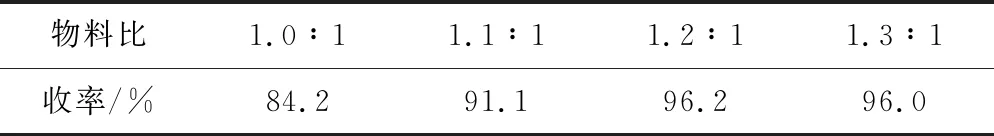
表2 物料比对中间体Ⅲ收率的影响
由表2可知,当物料比为1.0∶1时,中间体Ⅲ收率为84.2%;提高乙酰丙酮用量至物料比为1.2∶1时,收率达到最高;而继续增加乙酰丙酮用量,收率变化不大。因此,确定适宜的物料比为1.2∶1。
2.3 反应时间对中间体Ⅲ收率的影响
在n(TEA)∶n(β-硝基苯乙烯)为2.0∶1、n(乙酰丙酮)∶n(β-硝基苯乙烯)为1.2∶1、反应温度为25 ℃的条件下,考察反应时间对中间体Ⅲ收率的影响,结果见表3。

表3 反应时间对中间体Ⅲ收率的影响
由表3可知,延长反应时间,收率明显升高,当反应时间为3 h时,收率达到最高,为96.2%;继续延长反应时间,收率反而略有下降。因此,确定适宜的反应时间为3 h。
2.4 中间体Ⅲ和目标化合物Ⅰ的1HNMR 分析(图3)

图3 中间体Ⅲ(a)和目标化合物Ⅰ(b)的核磁共振氢谱
中间体Ⅲ的1HNMR分析:δ7.35~7.29,多重峰,积分3H,为苯环结构上的氢;δ7.20,双重峰,积分2H,为苯环结构上的氢;δ4.68~4.61,多重峰,积分2H,为硝基相连的亚甲基氢;δ4.39~4.37,多重峰,积分1H,为羰基α位次甲基氢;δ4.28~4.22,为硝基β位次甲基氢;δ2.30、δ1.94,均为单峰,积分3H,均为乙酰基上的甲基氢。
目标化合物Ⅰ的1HNMR 分析:δ11.01,宽峰,积分1H,为吡咯环NH上的氢;δ7.33~7.27,多重峰,积分3H,δ7.22~7.20,多重峰,积分2H,均为苯环结构上的氢;δ6.62,单峰,积分1H,为吡咯环5位碳上的氢;δ2.37,单峰,积分3H,为乙酰基上的甲基氢;δ1.93,单峰,积分3H,为吡咯环2位上取代的甲基氢。
3 结论
以β-硝基苯乙烯和乙酰丙酮为原料,在三乙胺碱性条件下发生Michael加成反应得到中间体3-(2-硝基-1-苯基乙基)戊烷-2,4-二酮,再经过硝基还原、关环反应得到目标化合物1-(2-甲基-4-苯基-1H-吡咯-3-基)乙酮,三步反应总收率为46.3%,中间体及目标化合物结构通过1HNMR 和MS表征。确定Michael加成反应的最佳条件为:碱为三乙胺、其用量n(三乙胺)∶n(β-硝基苯乙烯)为2.0∶1、物料比n(乙酰丙酮)∶n(β-硝基苯乙烯)为1.2∶1、反应温度25 ℃、反应时间3 h,在该条件下,Michael加成产物中间体Ⅲ收率为96.2%。该合成路线对于2,3,4-取代的吡咯类化合物的合成具有一定的指导意义。
猜你喜欢
佳木斯大学学报(自然科学版)(2022年1期)2022-11-25
云南化工(2022年9期)2022-10-12
中国药学药品知识仓库(2022年10期)2022-05-29
化学工业与工程(2022年1期)2022-03-29
材料科学与工艺(2022年1期)2022-03-11
能源化工(2021年6期)2021-02-26
汕头大学学报(自然科学版)(2020年4期)2020-12-14
生物工程学报(2020年1期)2020-03-12
现代农业科技(2018年23期)2018-02-18
大众科学(2017年10期)2017-12-18
