
癌症单细胞数据拷贝数变异检测方法
2021-06-23 01:35徐安琪高敬阳
北京化工大学学报(自然科学版) 2021年3期
徐安琪 蔡 磊 高敬阳
(北京化工大学 信息科学与技术学院, 北京 100029)
引 言
拷贝数变异(copy number variation, CNV)是人类基因组中一种重要的结构变异(structural variation,SV),它的位点突变率远高于单核苷酸变异(single nucleotide polymorphism,SNP)[1-2], 是人类疾病的重要致病因素之一。多项研究表明,拷贝数变异与多种人类肿瘤的发生密切相关,例如乳腺癌、肝癌、肾癌、食管癌等[3-5]。
现有的二代/三代测序技术使用的测序数据是多种细胞的混合样本,对这样的数据进行分析得到的结果只是一群细胞中信号的平均值,单个细胞的特异性被忽略,导致细胞的异质性不能得到体现[6],而异质性恰恰是癌症发生、发展和复发的基础[7]。作为在单个细胞水平上对基因组、转录组、表观组进行高通量测序以揭示单个细胞的基因结构和基因表达状态的新技术,单细胞测序(single cell sequencing,SCS)是研究细胞异质性的有力工具[8],对于探索癌症细胞异质性有着重要的意义。但由于单细胞测序是在单个细胞水平上进行的测序,所以需要对单个细胞微量全基因组DNA进行扩增。一般使用全基因组扩增(whole genome amplification,WGA)技术进行扩增,这种扩增技术可以大幅度增加DNA总量,但同时也会使得到的数据具有较低的基因覆盖度和较高的扩增偏好性,从而导致定量分析结果不准确[9]。
对于这种具有低覆盖度和高扩增偏好性的单细胞数据,大多数用于高通量测序(next generation sequencing,NGS)的CNV检测方法并不能取得良好的检测效果,只有少数可适用于单细胞数据CNV检测,这些方法从原理上基本可以分为3类,即滑动窗口、构造函数和隐马尔可夫模型[10]。这3种方法各有其优势和局限性[11-12]:滑动窗口能合并相同状态区间以提高断点检测的准确性,但忽略了单细胞数据覆盖度低和分布不均衡的数据特点,导致产生大量假阳性变异;构造函数虽然能很好地适应单细胞的数据特征,但对变异区间和断点的检测不考虑区域性,无法很好地合并公共断点; 隐马尔可夫模型虽然擅长处理异常数据点,但易出现过拟合或拟合程度不高的情况。
目前已有的这些方法各有利弊,且由于单细胞数据的特异性从而普遍存在检测精度不高、变异类型识别不准确、假阳性变异数量偏高等问题[13]。为了提高检测的精度,减少单细胞数据CNV检测中的假阳性变异结果,本文提出了一种针对单细胞测序数据的拷贝数变异检测方法(FL- CNV),通过对不同检测方法的优势进行互补,以期解决现有检测中遇到的精度问题和假阳性问题,并在后续实验中验证了本文方法用于单细胞CNV检测时精度的提高。
1 拷贝数变异检测方法
1.1 工作流程
FL- CNV工作流程如图1所示。首先该方法综合了冷泉港实验室不定长窗口的数据预处理思想[10],以避免由于删除低映射质量序列而出现的假阳性变异;其次利用构造函数方法中负二项建模的降噪优势进行数据处理,以修正在GC矫正中不能有效消除的数据偏好性;最后通过计算单细胞测序数据降噪后的当前窗口及周边一定数量窗口的读段深度(read depth,RD)数据[14],来确定数据异常范围并得到断点的位置及变异类型。确定断点和变异类型的步骤执行两次,第一次粗略划定可能存在变异的区域,第二次进一步细化最终的变异区间。
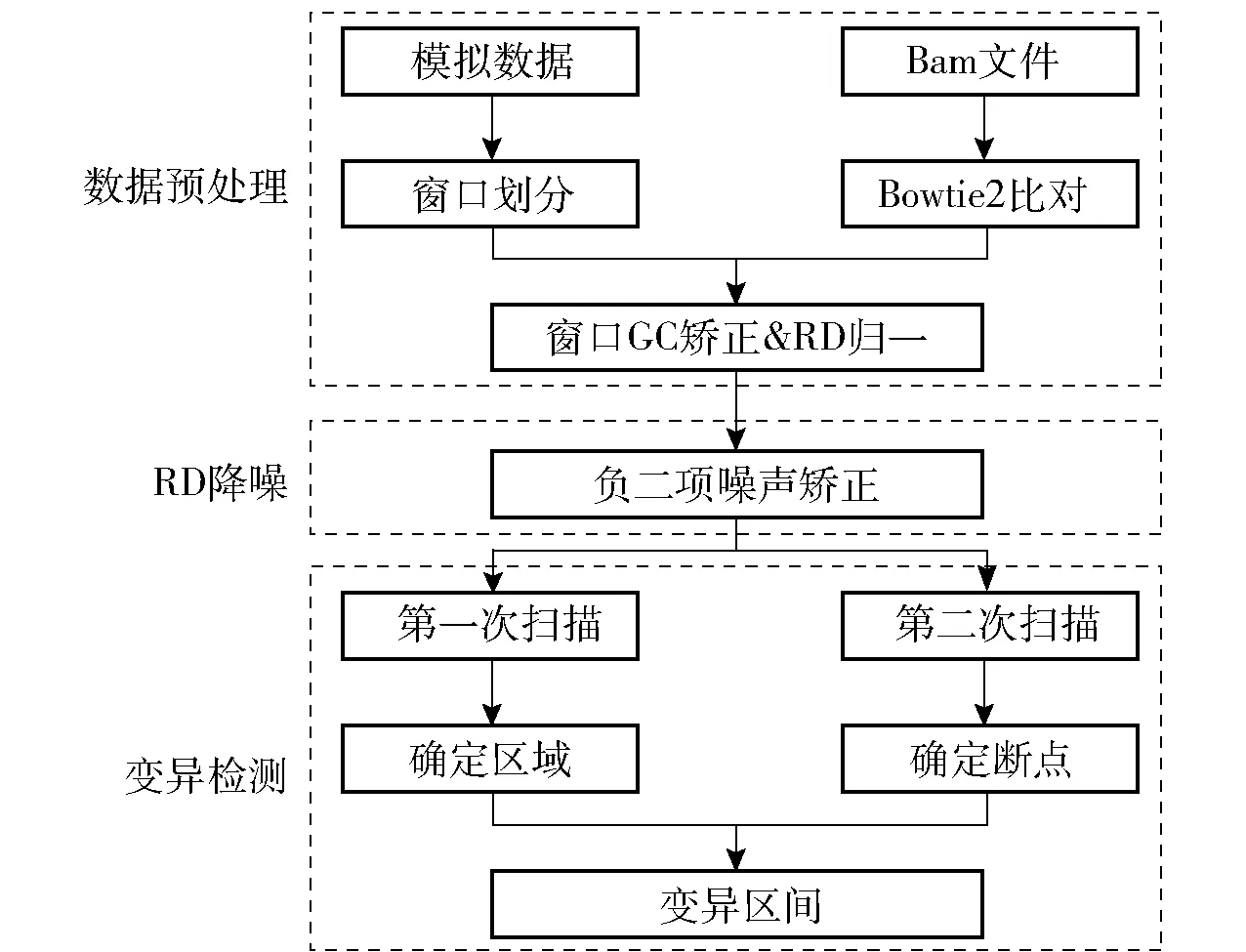
图1 FL- CNV工作流程Fig.1 The workflow for FL- CNV
检测流程如下:1)数据预处理,使用模拟数据将参考基因分割成变长窗口,通过Bowtie2将单细胞reads比对到参考基因,统计窗口真实数据RD,对窗口RD进行GC矫正;2)RD降噪,对步骤1)输出的RD数据进行矫正,消除扩增带来的噪声;3)变异检测,综合当前窗口与前后相邻若干窗口的RD值,通过两次窗口扫描确定变异。
1.2 数据预处理
数据预处理的步骤如下。
(1)窗口划分 为了避免单细胞数据覆盖率低且分布不均匀的特性对变异检测的影响,采用可变长的窗口来划分参考基因。首先以参考基因hg19为模版使用art_illumina软件[15]生成数据预处理阶段所需的模拟数据,再将模拟数据比对回参考基因(此处使用比对工具Bowtie2,参数设置与后续的真实数据比对相同),根据比对回参考基因的模拟读数的分布,将染色体划分为具有相同数量的模拟读数的窗口,即窗口划分规则是每个窗口内比对的模拟读数相同。已有研究表明窗口划分的最适数量为50 000左右[11],故设定比对到每个窗口的模拟读数数量为3 621,将全基因划分为50 042个窗口。因为单细胞数据分布不均衡,所以划分出的窗口长短不一,格式如表1所示,其中,Chr为窗口所在染色体号,Start为窗口的起始位置,End为窗口终止位置,Size为窗口大小,Reads为落在该窗口的reads数。
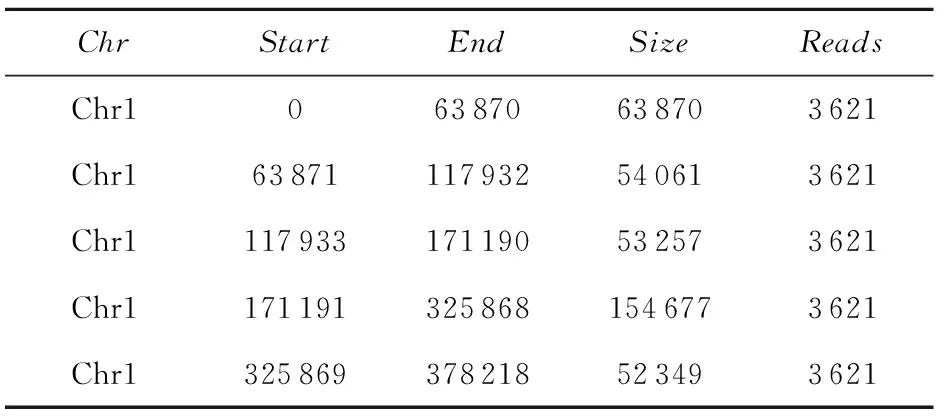
表1 窗口文件Table 1 Window file
(2)比对得窗口读段数量(read count,RC) 使用Bowtie2将单细胞的真实数据比对到参考基因,将窗口文件转化为bed文件,将bed文件和比对后得到的Bam文件作为输入,通过bedtools计算得到每个窗口的RC及GC含量。
(3)GC矫正 单细胞数据不均衡性的主要来源是整个基因组中GC含量的变化,为了对此进行调整,使用deeptools根据GC含量对窗口内的RC进行矫正。
(4)归一化 将GC矫正后得到的RC通过归一化得到RD。
1.3 负二项分布消除RD噪声
仅使用GC矫正不足以消除WGA带来的偏差,还需要进一步的矫正。常用的矫正方式有两种:第一种是设置对照组,将对照组样本测序数据作为参考,对每个窗口进行矫正;第二种是通过分析噪声特点有针对性地消除噪声。已有研究表明,来自单细胞测序数据的RD信号的固有噪声可以通过负二项(NB)分布准确地表征[11]。现有的利用负二项分布建模的方式虽然在确定断点和变异类型时效率低下,但在消除RD噪声方面性能良好,对比之下选择使用负二项分布对窗口RD进行矫正。
1.4 变异检测
构造函数具有良好的数据矫正能力,但其对断点的判断缺乏全局性考量,在变异区域的融合和共享断点的合并上存在明显缺陷。为达到更好的边界检测效果,本文摒弃通过目标函数求导确定断点的方法,通过两次扫描全局窗口的线性计算,高效准确地划分可能存在变异的区域,具体做法如图2所示,步骤如下。
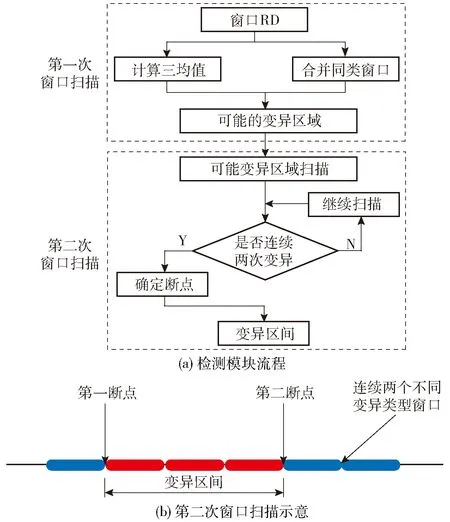
图2 变异检测模块Fig.2 Variation detection module
(1)第一次窗口扫描 从头依次扫描每个窗口,计算该窗口及其前10个窗口和后10个窗口矫正后RD值的三均值M3,若M3落在正常阈值外,则认为当前窗口内可能存在发生拷贝数变异的区域。这里所述的正常阈值范围为(N-N·m·n,N+N·m·n),其中N为样本倍性,n为所有窗口数据的标准差,m取1、2或3。在依次扫描全部窗口后,将变异类型相同的窗口标记为一个变异区域,初步得到可能存在变异的区域。
这里使用的三均值是概率分布中的概念,计算公式如下。
M3=Q1/4+Q2/2+Q3/4
(1)
式中Q1、Q3为数据的两个四分位点,Q2为中位数。三均值是一个有25%崩溃点的具有统计学抗性的L-估计,既对当前窗口进行了估计,又考虑到全局特性并排除了总体中的异常数值。
(2)第二次窗口扫描 以第一次划分的某个变异区域为例,首先从变异区域的第一个窗口开始,计算该窗口及其前后5个窗口RD值的平均值S2,定义第一个S2落在正常阈值外的窗口为第一断点,依次向后扫描计算,当出现连续两个窗口变异类型与之前窗口的不同时,定义出现变异类型转换的窗口的前一个窗口为第二断点。这样对某个初次划分的可能存在变异的区域进行二次扫描后,将其粗略估计的变异区域进一步细化,得到最后的断点和变异区间,如图2(b)所示。
2 实验及结果分析
为了验证本文方法的优越性,通过仿真数据实验和真实数据实验来评估方法的检测效果。首先比较了FL- CNV与nbCNV[11]、control-FREEC[16]、CNVnator[17]在SCSsim[18]生成的模拟数据上的性能,然后在100个导管癌的真实数据上以倍体信息及aCGH的拷贝数划分作为方法性能的真实性验证[19]。
2.1 模拟数据实验
2.1.1数据生成
在模拟实验方面,本文选取了千人基因组的人类hg19参考基因的21号染色体为模版,人为地随机引入拷贝数变异事件。由于单细胞噪声问题突出,所以有效地模拟单细胞的数据特征对于单细胞测序工具的基准测试来说是非常必要的。不同于数据预处理阶段,在模拟实验阶段本文不再使用传统仿真工具art_illumina,而是选择专用于生成单细胞测序数据的仿真工具SCSsim来生成模拟实验所需的数据。
模拟数据的生成分为3步:首先通过SCSsim的simuvars功能定义拷贝数的变异位点及变异类型,每次模拟实验都随机引入15个互不重叠的拷贝数变异事件生成fasta文件;之后基于由Illumina仪器生成的正常细胞的实际测序数据推断测序概况,构建profile配置文件,此处使用导管癌T10的一个单细胞数据,即索引号为SRR052047的单细胞测序样本;最后基于fasta文件及profile文件得到fastq格式的序列片段文件。
2.1.2评价指标
模拟实验通过精度Pre、敏感度Sen和F1系数来评估每个方法的性能,各参数的计算公式如下。
Pre=TP/(TP+FP)
(2)
Sen=TP/(TP+FN)
(3)
F1=(2Pre·Sen)/(Pre+Sen)
(4)
式中,TP表示真阳性,FP表示假阳性,FN表示假阴性。
2.1.3实验结果
本文进行了100次模拟实验,共获得了1 500个拷贝数基准变异,使用FL- CNV、control-FREEC、nbCNV和CNVnator这4种方法对模拟数据进行变异检测,结果如表2所示。
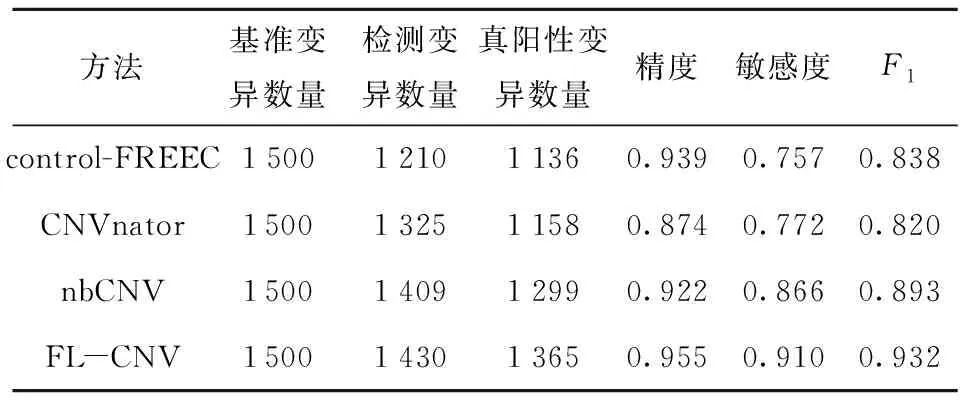
表2 模拟数据实验结果Table 2 Experimental results with simulated data
由表2中数据可知,FL- CNV在模拟实验中表现良好,检测精度明显高于其他3种方法,与此同时敏感度也是4种方法里最高的。可见本文方法在保证不损失敏感度的同时,显著提高了检测的精度,达到了较好的检测效果。
2.2 真实数据实验
由于单细胞缺少CNV基准数据,所以现有的单细胞CNV检测方法在进行性能评估时,对方法检测效果的验证大都使用倍体信息作为类标签对细胞个体数据进行聚类分析,但这样的验证方法并不能直观地体现变异检测效果,因为倍体信息仅代表了样本序列拷贝数的平均概况,并不能量化单个细胞的变异检测水平。为了更直观地比较变异检测效果,在真实数据实验部分,我们不但使用真实数据的倍体信息来衡量方法的好坏,还将Navin等[10]提出的比较基因组杂交的拷贝数划分作为金标准来直观比较方法性能。
2.2.1数据概况
真实数据为来自美国国家生物技术信息中心(National Center of Biotechnology Information, NCBI)的100个导管癌(T10)单细胞,编号为SRP002535(https:∥www.ncbi.nlm.nih.gov/sra/?term=SRP002535)。该数据集有47个二倍体D,24个亚二倍体H和29个异倍体,其中异倍体又可分为异倍体AA(3N)和异倍体AB(3.3N)。用于检测效果验证的比较基因组杂交(array-based comparative genomic hybridization,aCGH)基准数据来自NCBI,编号为GSE16607。
2.2.2实验结果
为了直观地比较不同方法的检测效果,将4种方法在这100个单细胞数据上的全基因拷贝数检测结果整理为4个矩阵,生成如图3所示的热图。其中每行代表一个细胞个体,每列代表一个检测窗口,游标为拷贝数与热图中每个窗口颜色的映射关系。
由图3可以看出,FL- CNV和nbCNV能将100个单细胞分为明显的3类,而control-FREEC与CNVnator对100个导管癌单细胞的聚类效果则不太理想,远不如FL- CNV和nbCNV的分类效果清晰明了。

图3 不同方法的热图Fig.3 Heatmap displays of different methods
为进一步直观比较其他检测方法与本文方法的优劣,选择在热图中表现良好的nbCNV与本文方法进行皮尔逊相关系数(Pearson correlation coefficient)的比较,结果如图4所示。由图4可以看到,综合不同亚群的相关性情况,FL- CNV整体来说具有比nbCNV更高的皮尔逊相关系数,即FL- CNV检测的拷贝数变异情况更符合真实数据的拷贝数变异情况,也即FL- CNV检测出的拷贝数变异具有更高的可信度。
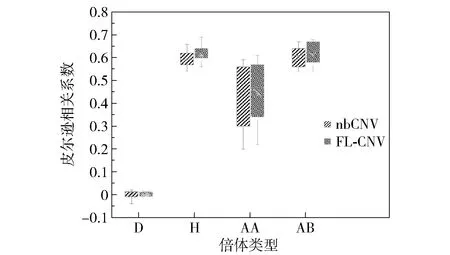
图4 皮尔逊相关系数比较Fig.4 Pearson correlation coefficient comparison
综合以上模拟数据和真实数据的实验结果可知,本文方法在用于拷贝数变异检测时具有更高的精度和敏感度;在真实数据实验部分,本文不仅通过热图定性地比较了不同方法对单细胞数据的分类能力,并且通过皮尔逊相关系数定量地比较了所提方法和nbCNV对单细胞拷贝数变异的检测能力,实验结果更加可靠。
3 结论
本文提出了一种基于单细胞测序数据的拷贝数变异检测方法FL- CNV,根据单细胞测序数据的特点,动态划分变长窗口并矫正窗口数据,对矫正后的窗口进行两次数据估算,这两次数据估算既顾及到全局数据概况又考虑了局部数据变化,有效地避免了异常数据点对变异检测带来的影响,减少假阳性样本的产生,提高了变异检测方法的精度和敏感度,解决了现有检测方法精度不高的问题。
模拟数据和真实数据的实验结果验证了本文方法的优越性,并且避免了单细胞实验验证因缺少基准数据而采用倍体标签来衡量方法好坏的不准确的结果检验方式。以比较基因组杂交的拷贝数划分作为金标准,通过热图和皮尔逊相关系数的比较进一步证明了本文所提FL- CNV具有优于其他检测方法的精度、敏感度和可信度。
猜你喜欢
中国癌症防治杂志(2022年4期)2022-11-24
中华骨与关节外科杂志(2022年1期)2022-08-31
学苑创造·C版(2022年8期)2022-06-18
辽河(2022年4期)2022-06-09
右江民族医学院学报(2022年2期)2022-05-19
教育界·上旬(2020年8期)2020-06-27
江苏农业科学(2019年14期)2019-09-23
电脑报(2019年20期)2019-09-10
初中生世界·九年级(2019年6期)2019-08-15
江苏农业科学(2017年16期)2017-10-27
