
Selective Conversion of CO2 by Single-Site Catalysts
2021-06-02 11:38XinjiangCuiFengShi
物理化学学报 2021年5期
Xinjiang Cui , Feng Shi
State Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China.
Abstract: Industrial revolution has led to increased combustion of fossil fuels.Consequently, large amounts of CO2 are emitted to the atmosphere, throwing the carbon cycle out of balance. Currently, the most effective method to reduce the CO2 concentration is direct CO2 capture from the atmosphere and pumping of the captured CO2 deep underground or into the mid-ocean. The transformation of CO2 into high-value chemicals is an attractive yet challenging task. In recent years, there has been much interest in the development of CO2 utilization technologies based on electrochemical CO2 reduction, photochemical CO2 reduction, and thermal CO2 reduction, and CO2 valorization has emerged as a hot research topic. In electrochemical CO2 reduction, the cathodic reaction is the reduction of CO2 to value-added chemicals. The anodic reaction should be the oxygen evolution reaction, and water is the only renewable and scalable source of electrons and protons in this reaction.There is a plethora of research on the use of various metals to catalyze this reaction. Among these, Cu-based materials have been demonstrated to show unique catalytic activity and stability for the electrochemical conversion of CO2 to valuable fuels and chemicals. Moreover, the solar-driven conversion of CO2 into value-added chemical fuels has attracted great attention, and much effort is being devoted to develop novel catalysts for the photoreduction of CO2, especially by mimicking the natural photosynthetic process. The key step in the photocatalytic process is the efficient generation of electron-hole pairs and separation of these charge carriers. The efficient separation of photoinduced charge carriers plays a crucial role in the final catalytic activity. Compared with CO2 reduction via electrocatalysis and photocatalysis, thermal reduction is more attractive because of its potential large-scale application in the industry. Heterogeneous nanomaterials show excellent activity in the electrocatalytic, photocatalytic, and thermal catalytic conversion of CO2. However,nanostructured materials have drawbacks on the investigation of the intrinsic activity of the active sites. In recent years,single-site catalysts have become popular because they allow for maximum utilization of the metal centers, show specific catalytic performance, and facilitate easy elucidation of the catalytic mechanism at the molecular level. Accordingly,numerous single-site catalysts were developed for CO2 reduction to produce value-added chemicals such as CO, CH4,CH3OH, formate, and C2+ products. Value-added chemicals have also been synthesized with the aid of amines and epoxides. This review summarizes recent state-of-the-art single-site catalysts and their application as heterogeneous catalysts for the electroreduction, photoreduction, and thermal reduction of CO2. In the discussion, we will highlight the structure-activity relationships for the catalytic conversion of CO2 with single-site catalysts.
Key Words: CO2 reduction; Single site catalyst; Carbonylation; Electrocatalysis; Photocatalysis;Thermal catalysis
1 Introduction
The CO2concentration increases year by year causing serious global warming problems1and being a major threat to sustainable human development2-4. To decrease the CO2emission, CO2capture is considered to be the most important step and represents a normal strategy to remove CO2from industrial waste gases5-9. To use the captured CO2, one of the most attractive approaches is to convert it into highly valuable chemicals10-15. Any technologies such as electrochemical,photochemical, biochemical or thermochemical, have been intensively explored on the sustainable transformation of CO2to value added chemicals15-18. To efficiently and selectively catalyzed the conversion of CO2to chemicals, catalysts with active metal centers are extremely necessary. To date, different types of catalysts such as homogeneous-, heterogeneous- and biological catalysts are widely developed19-29. Compared with their homogeneous and biological counterparts, heterogeneous catalysts have intrinsic advantages such as high stability, easy separation from the reactants and products, practical operation under harsh conditions and large scale industrial applications30,31.Although these advantages, limited information is available on the catalytically active sites in traditional heterogeneous supported catalysts.
Recently, single site catalysts (SSCs) where individual metal atoms are stabilized by covalent coordination or ionic interactions with neighboring surface atoms have attracted extensive attention in the fields of heterogeneous catalysis32-36.Moreover, the catalytic performance of SSCs strongly depends on the coordination environment between the central metal and the neighboring atoms1,37,38. Similar to molecularly-defined homogeneous catalysts, single site catalysts display unique electronic properties and allow for metal utilization up to 100%.Thus, these atomically dispersed metal catalysts can offer new opportunities to investigate the catalytic mechanism on a molecular level39,40. Based on these inherent advantages,growing efforts have been made to apply SSCs on selective CO2transformation, as shown in Fig. 1.
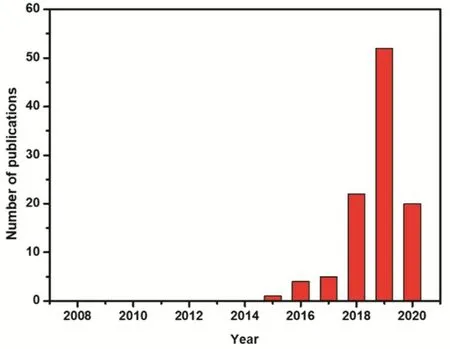
Fig. 1 Number of publications using the terms “Single Atom” or“Single Site” and “Carbon Dioxide” or “CO2” from 2007 to May of 2020 by the ISI web of science.
In this review, we will focus on recent significant achievements in selective transformation of CO2applying SSCs.We wish to provide a comprehensive understanding on how the individual single sites can improve the catalytic performance and motivate the developments of selective CO2reduction. Finally,we outline the remaining challenges and opportunities in this field.
2 Catalytic electroreduction of CO2
In general, the catalytic electronic reduction of CO2is a multistep reaction that a proton-assisted process occurs at the cathodes by transferring multiple-electrons. Because different electrons or protons can be transferred during the reaction, CO2can be converted into different kinds of valuable chemicals, including C1products (carbon monoxide, methane, methanol, formic acid,formaldehyde)41-44, and C2+products (ethylene, ethane, ethanol,oxalic acid)45-49. In this part, we will summarize recent advances on the catalytic electroreduction of CO2by SSCs.
2.1 C1 chemicals synthesis
2.1.1 Nickel catalysts
Li and coworkers fabricated a Ni single site catalyst where the single Ni centers were immobilzed on N-doped porous carbon(Ni/N-C) by ionic exchange of Zn nodes and adsorbed Ni salts(Fig. 2a-c). Compared with Ni nanoparticles (NPs), Ni SSCs are capable of selectively reducing CO2with excellent current density and a Faradaic efficiency (FE) for CO production (FECO)of over 71.9%. In contrast to Ni NPs, the Ni SSCs displayed a lower interfacial charge-transfer resistance favoring the faster electron transfer from the electrodes to CO2and easier formation of theradical anion intermediate. These results indicated a different mechanism for the reduction of CO250.

Fig. 2 (a, b) Magnified HAADF-STEM images of Ni SAs/N-C.The Ni single atoms are marked with red circles. (c) Corresponding EDS maps revealing the homogeneous distribution of Ni and N on the carbon support. (d) STEM-EDS mapping of Ni in Ni/N-CNTs.(e) AC-STEM-annular dark-field (ADF) images showing the atomic dispersion of Ni in Ni/N-CNTs. (f) ADF image showing the Ni single atoms located on the walls of a CNT (The red circles show typical Ni atoms embedded in the carbon plane of walls. Color online).
Jiang and coworkers synthesized a nickel SSCs by a novel multistep pyrolysis steps. Using this novel process, single nickel sites loading as high as 20% (w, mass fraction) was obtained by coordinating with N to form Ni-Nxspecies in the N-CNTs (Ni/NCNTs, Fig. 2d-f). The formed Ni-SSCs exhibited outstanding activity on the electrochemical reduction of CO2to CO compared with its Ni-NPs counterparts on N-CNTs. During the multistep pyrolysis process, the Ni-O-C on the melem skeleton was formed firstly by the condensation of the dicyandiamide at ≈ 350 °C. Consequently, Ni single sites was supported on γ-C3N4at 650 °C by strong covalent bonding between the Ni atoms and γ-C3N4attributing to the high loading of Ni51.
Later on, the individual Ni atoms homogeneously dispersed on the graphene oxide (GO) sheets were prepared. The isolated nickel ions are stabilized by the strong interactions with the GO and the tris(2-benzimidazolyl-methyl)amine (NTB) linker forming Ni(NTB)-GO. After pyrolysis of Ni(NTB)-GO under high-temperature, the isolated nickel sites were supported on the surface of the nitrogen-doped graphene oxide sheets. The formed Ni-N-RGO shows high CO2reduction selectivity in the reduction of CO2to CO with 97% FE at -0.8 V. In this case, the NiN4was proved to be the active sites52. Wang and coworkers fabricated SSCs where the Ni single atoms are homogeneously dispersed into graphene nanosheets. The Ni single sites acted as active sites for the electrocatalytic CO2conversion to CO. These single atomic Ni sites displayed a high 95% selectivity of CO with an excellent stability over 20 hours’ continuous electrolysis.When scaling the current density up to 50 mA·cm-2, 97% selectivity of CO remained53.
Low-valent Ni(I) single atoms were successfully anchored on N-doped graphene matrix. The resulting catalyst acted as a robust and efficient electrocatalyst for CO2reduction with high activity and stability. X-ray absorption spectroscopy (XAS)study revealed that the unpaired electron was delocalized in the Ni3dx2-y2orbital and the charges was spontaneously transferred from Ni(I) to the carbon 2p orbital in CO2formingspecies.The formation ofspecies was favored the reduction the energy barrier for electroreduction of CO2. The high activity of the electroreduction of CO2are ascribed to the fabrication of a single-atom low-valent Ni(I) heterogeneous catalyst and the active site geometry and structural transitions during CO2reduction to CO54.
Liu and coworkers reported a strategy for the synthesis of Ni SSCs by the very simple impregnation-pyrolysis of graphene oxide aerogel with nickel salts. The resulting materials showed a three dimensional structure where the single Ni sites were coordinated with the N-species formed during the pyrolysis. The Ni-SSCs exhibited outstanding activity toward electroreduction of CO2to CO with a remarkable CO FE of 90.2%. DFT study disclosed that the intermediate *COOH which favored to generate CO was preferred to be formed on coordinatively unsaturated Ni-N sites with lower free energies than that on Ni-N4site, demonstrating the excellent activities of CO2electroreduction in this Ni-SSCs55. Moreover, a Ni single-atom catalyst which is synthesized from a novel metal-organic complex precursor [Zn(Ni)-bidppz] [11,11’bis(dipyrido -[3,2-a:2',3'-c]phenazinyl) = bidppz] exhibited significantly enhanced performances for CO2conversion. Compared with a pure N-C sample, this Ni-SSCs offered high FECOof ~91.2% and 2.15-fold improvement in current density at -0.9 V56.
Yu and coworkers reported that the Ni SSCs can be fabricated by pyrolysis of a bimetallic metal-organic framework (MgNi-MOF-74). The introduction of polypyrrole (PPy) unit in MgNi-MOF-74 matrix offered the N source to stabilize the isolated Ni atoms during pyrolysis meanwhile the large amount of Mg2+helped to extend the spatial isolation of Ni2+. Interestingly, a series of single atom Ni catalysts (named Ni-SSC-Nx-C) with different N coordination numbers could be easily manipulated by changing the pyrolysis temperature. the (Fig. 3a-c). After optimization, the Ni-SSC-N2-C catalyst, with the lowest N coordination number, displayed the highest CO FE (98%) and turnover frequency (1622 h-1) of electroreduction of catalytic CO2to CO compared with that of Ni-SSC-N3-C and Ni-SSC-N4-C (Fig. 3d,e). DFT calculations was conducted to investigate the mechanism and revealed that the intermediate of COOH* was favorably formed by the Ni-SSC with low N coordination number leading to the superior activity towards CO57.
Lu and coworkers demonstrated a practical strategy to prepare Ni-SSC from nickel chloride (NiCl2), cyanuric acid (CA) and 2,4-diamino-6-phenyl-1,3,5-triazine (DPT). As shown in Fig.4a, after immobilizing on carbon cloth and heating treatment under 500 °C, N/O mixing coordinated Ni-N3O-SSC could be synthesized by the reaction of CA and DPT. Because of the weaker Ni-O interaction, the oxygen atom can be gradually removed under 800 °C generating a vacancy-defect Ni-N3-V SSC (Fig. 4b,d). N2 sorption isotherm showed the pore diameter distributions with the size of ~1 nm, Fig. 4c. Compared with the vacancy-defect free Ni-N4-SSCs which is synthesized from Ni(II) precursor and nitrogen containing ligand, the introduction of vacancy-defects in Ni-N3-V-SSC can significantly improve the electroreduction of CO2to CO under current density of 65 mA·cm-2with high Faradaic efficiency (FE) over 90% at 0.9 V vs reversible hydrogen electrode (RHE)58.
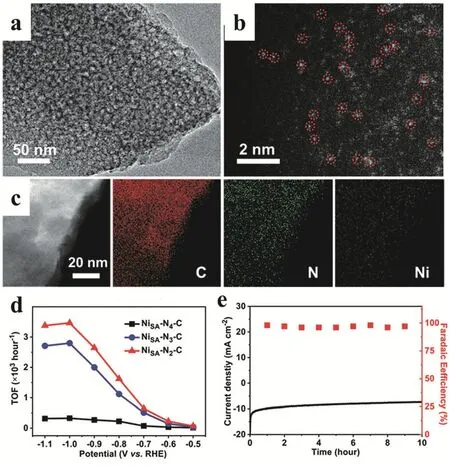
Fig. 3 (a) TEM image and (b) Aberration-corrected high-angle annular dark field scanning transmission electron microscopy(HAADF-STEM) image of Ni-SSC-N2-C. (c) EDS mapping of Ni, N and C elements in Ni-SSC-N2-C. (d) FEs of CO at different applied potentials and (e) stability of Ni-SSC-N2-C at -0.8 V during 10 h.
Liu prepared a Ni-SSCs by a two-step approach where the well-defined molecular of nickel(II)-2,9,16,23-tetra-(amino)phthalocyanine was synthesized and then linked to a conductive carbon support. The developed single Ni sites had a uniform structure with well-defined Ni-N4moieties and exhibited high catalytic performance on the conversion CO2to CO with FE of 99% and TOF of 100.179 h-1at a current density of 32.3 mA·cm-2and over-potential of 600 mV. Operando spectroscopic and electrochemical kinetics studies revealed that the in-situ formation of Ni+by the reduction of Ni2+was the highly active sites during the electroreduction of CO2. Further investigations confirmed that the rate-determining step of the CO2reduction was *CO2+ H+→ *COOH59.
It is still a challenge to prepare the industrial scale electrodes.To achieve this goal, the catalysts are fixed on a conductive substrate with the help of insulating polymer binders, such as Nafion or PVDF. This protocol largely increases the synthetic complexity and cost. Thus it is highly desirable to develop a strategy to prepare the electro-catalysts in large scale. Recently,Li and coworkers demonstrated a method to prepare synthesize hierarchical, self-supported, and atomistic nickel catalyst by the solid-state diffusion between the N-doped carbon phase and bulk Ni metal (Fig. 5a,b). The resulting Ni catalyst was programmable and scalable prepared satisfying the industrial demand and was directly used as a binder-free electrode on the electroreduction of the CO2to CO under a current density of 48.66 mA·cm-2at 1.0 V versus RHE with high faradic efficiency of 97% to CO. Interestingly, over 90% of selectivity was retained in a wide working potential of 0.7 to 1.2 V. This solid-state diffusion method offers a potential strategy to produce atomic catalysts industrially60. Moreover, Wang and coworkers synthesized an isolated Ni sties which supported on carbon black particles by a simple and scalable method. The resulting Ni-SSCs displayed an excellent performance for CO2electroreduction in a traditional H-cell with a CO Faradic efficiency of 99% in a KHCO3aqueous solution. More importantly, nearly 100% CO was produced in large current densities of 100 mA·cm-2showing 10-fold higher than the current densities in H-cell (Fig. 5c)61.
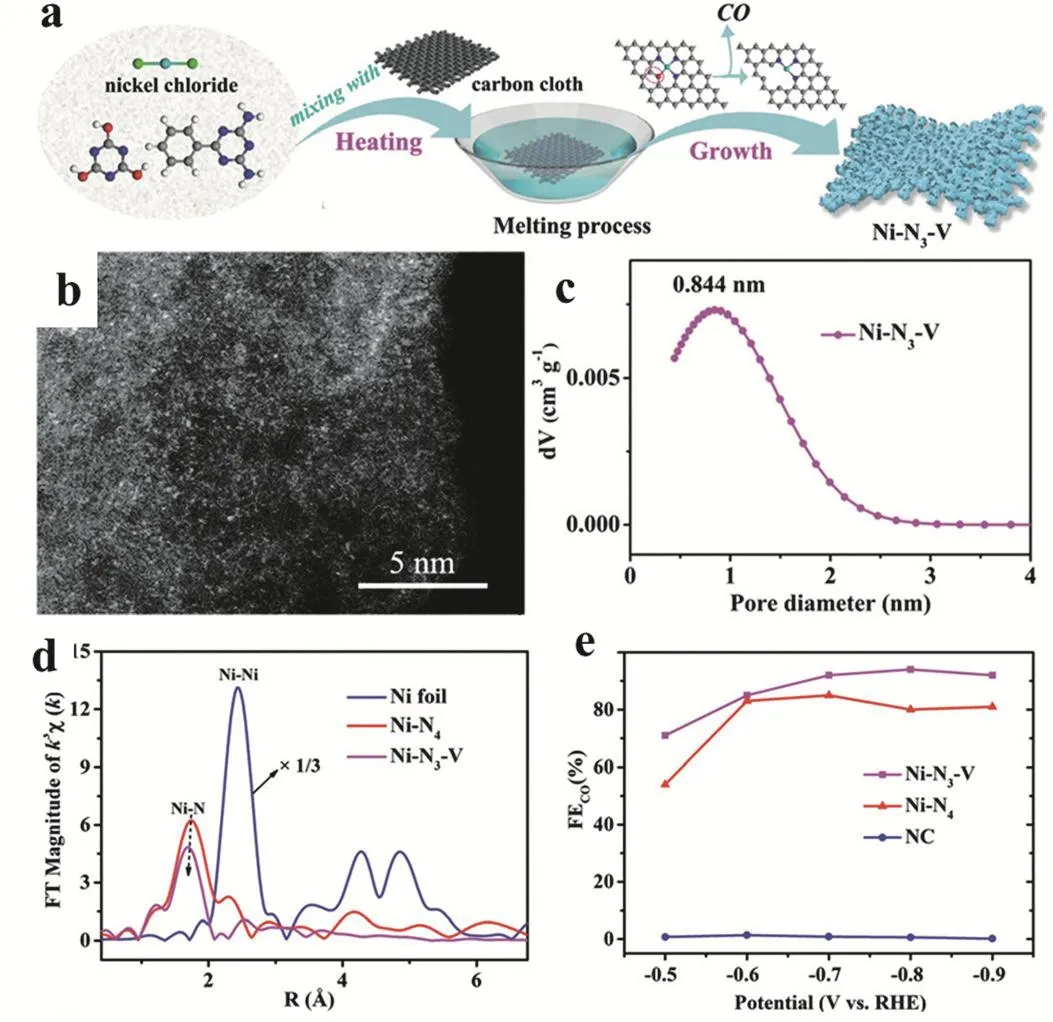
Fig. 4 (a) Illustration for the synthesis of Ni-N3-V. (b) HAADF-STEM image for Ni single atoms (bright dots) in Ni-N3-V. (c) The pore diameter distribution for Ni-N3-V obtained by nitrogen adsorption isotherm. (d) Ni K-edge k3-weighted FT-EXAFS spectra of Ni-N3-V,Ni-N4, and Ni foil (the EXAFS intensity of Ni foil is shown at one third value). (e) Specific current density of CO for Ni-N3-V, Ni-N4 and NC.
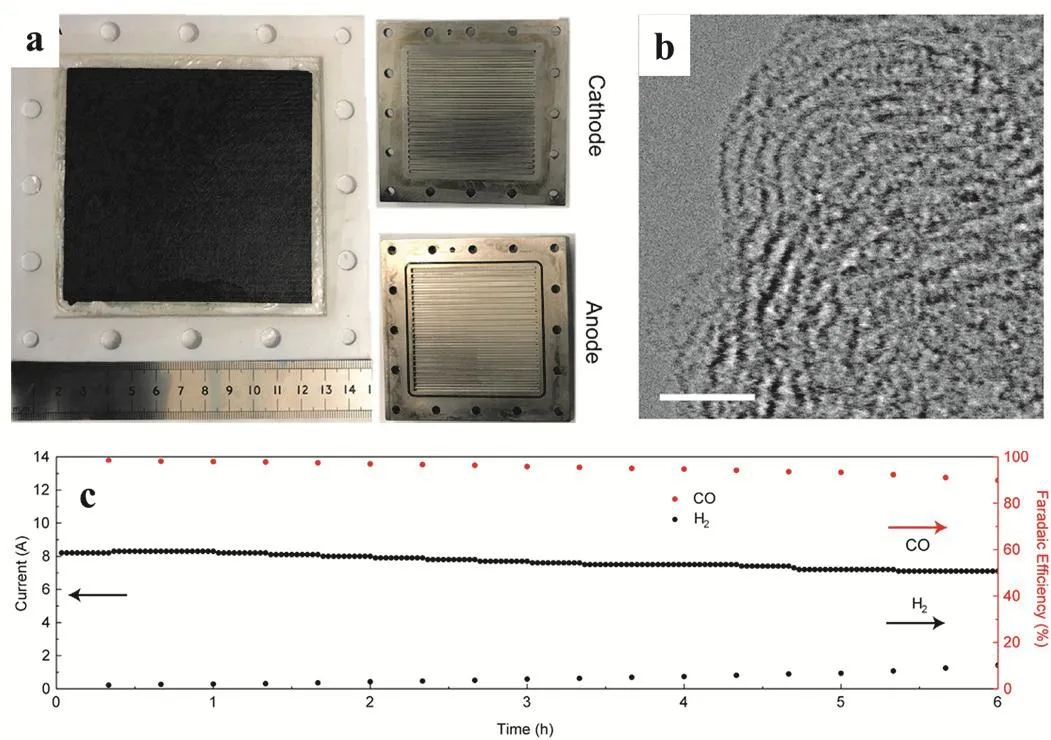
Fig. 5 (a) Photographs of assembled reactor, membrane electrode assembly, and individual cell components. (b) Aberration-corrected HAADF-STEM image. Scale bar represents 2 nm. Adapted from Ref. 60, Copyright 2019, Elsevier Inc. (c) The CO2RR stability test of Ni-NCB on CFP (1.25 mg·cm-2) in an H-cell under 0.55 V over-potential.
2.1.2 Cobalt catalysts
Beside the Ni-SSCs, Li and coworkers described a robust CO2reduction reaction electrocatalyst with atomically dispersed Co-N5sites. The Co-SSCs was synthesized by constructing coordination interaction between Co and N-rich materials(MRFPSs). The MRFPSs was synthesized firstly by the pyrolysis of core@shell materials of SiO2@melamineresorcinol-formaldehyde polymer spheres (MRFPSs) and etching the silica afterwards. The resulting Co-N5SSCs exhibited high selectivity for electroreduction of CO2with CO Faradaic efficiency (FE) over 90% in a wide range of potential from -0.57 to -0.88 V. Both experiments study and DFT calculations demonstrated the single-atom Co-N5sites are the active centers for COOH* formation as well as the CO desorption. Further investigation revealed that Co-N5sites exhibited a strong interaction with COOH and moderate interaction with CO making it an ideal catalyst for CO2reduction(Fig. 6a,b)62. Later on, the same group prepared series of Co-SSCs with various nitrogen coordination atoms and discovered the correlation of the coordination numbers with their CO2electroreduction performance. After testing, single Co site coordinated with two nitrogen atoms (Co-N2) displayed the superior activity affording both higher selectivity and activity with 94% of CO FE compared with that by Co-N3, Co-N4and Co nanoparticles (Fig. 6c,d)63.
The single site catalyst can afford an excellent platform to investigate the structure activity relationship. Tang and coworkers fabricated a well-defined Co SSCs on ultrathin nanosheets (STPyP-Co) by axial coordination assembly of a molecular tetra(4-pyridyl) porphyrin cobalt(II) (MTPyP-Co).The as-assembled catalyst displayed the precise identification of the Co sites for d-orbital energy engineering and showed excellent catalytic performance for the electroreduction of CO2with a remarkable CO FE of 96% at the overpotential of 500 mV(Fig. 7a,b). More importantly, the Co-SSC exhibited good stability for 48 h with a turnover frequency of 2.1 s-1. Structure activity relationship study revealed that the elevated dz2energy level in Co 3d orbital of Co-SSCs was ascribed to be the origin of its excellent electro-catalytic activity toward CO2reduction(Fig. 7c-f)64.
2.1.3 Cobalt-nickel bimetallic catalysts
Considering Co and Ni single-site catalysts are selective in producing H2and CO, respectively, it is highly desirable to fabricate electrocatalysts containing both Co and Ni. Chen and coworker synthesized such a catalyst (CoNi-NC) which exhibited a high syngas evolution capacity with total current over 74 mA·cm-2(Fig. 8a,b). The CO/H2ratios were in the range of 0.23-2.26, which are appropriate for the downstream applications (Fig. 8c). Moreover, CoNi-NC exhibited excellent electro-catalytic stability for 7 h (Fig. 8d). DFT calculations indicated that Co and Ni single sites were high active for the formation of the key reaction intermediates of *H, *HOCO and CO facilitating the HER and CO2RR, respectively. Moreover,the ratios of H2to CO could be tuned by varying the ratios of Ni to Co65.
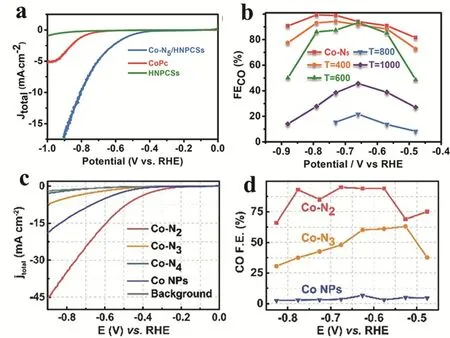
Fig. 6 (a) LSV curves of Co-N5/HNPCSs-T. (b) CO Faradaic efficiencies of Co-N5/HNPCSs-T Adapted from Ref. 62, Copyright 2018,American Chemical Society. (c) LSV of Co-N2, Co-N3, Co-N4, Co NPs and pure carbon paper as background.(d) CO Faradaic efficiencies at different applied potentials.
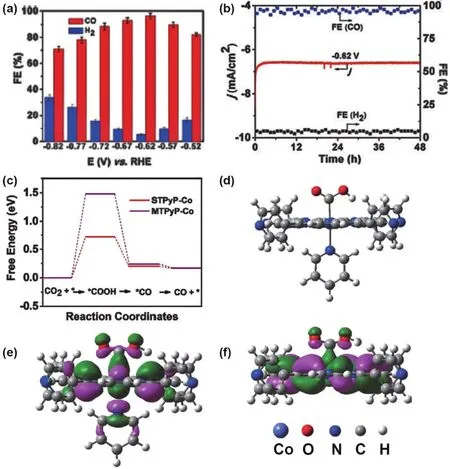
Fig. 7 (a) CO and H2 Faradaic efficiency (FE) for STPyP-Co at different applied potential. (b) Chronoamperograms and Faradaic efficiency(FE) of CO and H2 for STPyP-Co at @0.62 V vs RHE. DFT calculation of electrocatalytic CO2 reduction on STPyPCo. (c) Calculated freeenergy states of CO2 reduction to CO on STPyPCo and MTPyP-Co. (d) Optimized geometry of intermediate [STPyP-Co-COOH].(e) and (f) Spatial representation of HOMO orbital of [STPyP-Co-COOH] and [MTPyP-Co-COOH] intermediates, respectively.
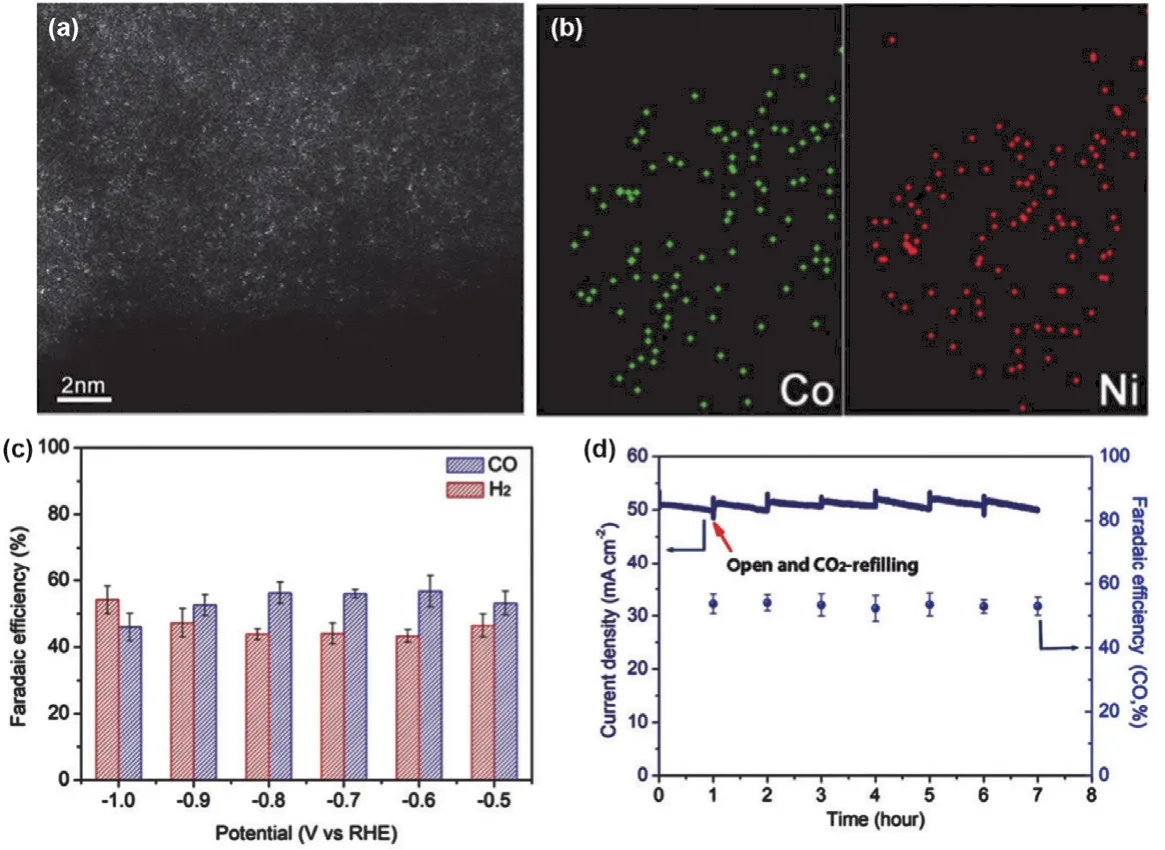
Fig. 8 (a) HAADFSTEM image of CONi-NC. (b) Elemental mapping of Co and Ni. (c) Faradaic efficiencies for CO and H2 evolution at different potentials. (d) Long-term stability measured at @0.9 VR.
2.1.4 Iron catalysts
Compared with other transition metals typically used in catalysis, iron has a number of advantages such as low price,non-toxicity and large abundance. However, it is challenging to fabricate and apply the Fe SSCs in the CO2transformation. Shao and coworkers synthesized a Fe SSCs employing the zeolitic imidazolate framework (ZIF-8) as a self-template. The isolated Fe sties were uniformly dispersed on N doped carbon. The resulting Fe-N-C model catalyst exhibited a high CO Faradaic efficiency (FE) of 93.5% under ultralow over-potential of 90 mV.The in situ Attenuated Total Reflectance Infrared Spectroscopy(ATR-IR) and DFT calculations revealed that the Fe centers in the defect-free graphitic layer were strongly adsorbed by *CO and hard to initiate the reduction of CO2-to-CO. Further investigation found that the synergistic interaction between the Fe-N4moiety and the defective graphitic layer possibly enhanced the CO2reduction reaction where the more balanced*COOH and *CO binding strength was achieved inhibiting the*CO poisoning. These results revealed that the hosting environments near the Fe centers can influence the catalytic performance significantly66.
Gascon and coworkers fabricated atomically dispersed iron sites on mesoporous nitrogen-doped carbon nanoparticles(mesoNCFe) by the pyrolysis of tetramethyl orthosilicate(TMOS) confined Fe-ZIF-8 MOF and studied its structure and catalytic performance relationship. The hydrolysis of TMOS in the MOF framework was occurred firstly and act as the key role to maintain a mesoporous property during the MOF hydrogenolysis favoring the formation of iron single sites.Compared with individual Fe on the microporous N-doped carbon (microNC-Fe), the mesoNC-Fe displayed CO production with the maximum FECOof 85% at -0.73 V because of the higher surface area of mesoNC (Fig. 9c). Combination of the experimental spectra and theoretical calculations disclosed the coordination of the isolated iron centers with porphyrinic species and OH/H2O moieties (Fe-Pc-OH-H2O) responsible for the high activity in CO2electroreduction (Fig. 9a,b,d). Fe-Pc-OH-H2O structure favored the CO formation because of the lower free energy barriers of *COOH and inhabitation of the *H67.
Wang reported a new approach for the synthesis of a robust single Fe site catalyst by thermal pyrolysis of graphene (G)adsorbing hemin (H) and melamine (M) (Fig. 10a,b). The resulting sample was consisted by dispersed FeN5sites and exhibited excellent electrochemical CO2reduction to CO with a high FE of ~97.0% at a very low over-potential of 0.35 V (Fig.10c). In FeN5, the coordination of the axial pyrrolic N species with isolated Fe centers reduced the electron density of Fe 3d orbitals and disfavored the Fe-CO π back-donation (Fig. 10d),leading to fast remove of CO and high selectivity of CO68.
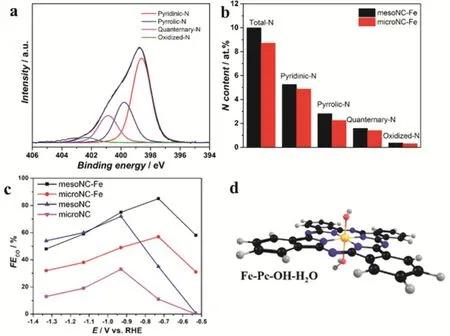
Fig. 9 (a) N 1s XPS regions of mesoNC-Fe with deconvolution into the N-speciation. (b) N distribution mesoNC-Fe and microNC-Fe.(c) FECO of mesoNC-Fe (black) microNC-Fe (red) mesoNC (blue) and microNC (pink) in CO2 reduction to CO. (d) DFT-optimized geometry for potential local environments of the iron active site in their most stable multiplicity state.
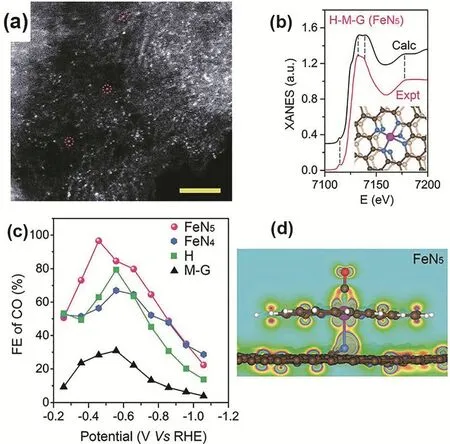
Fig. 10 (a) Magnified view of STEM images of uniformly distributed single Fe atoms in graphene, scale bar 2 nm. (b) The experimental XANES curves with FeN5 (pyrolyzed sample of hemin and melamine on graphene, H-M-G). (c) Faradaic efficiency (FE) of the electrocatalytic activity of as-synthesized catalysts. (d) Partial charge density of the plane formed by O-C-Fe-N-pyrrolic N within the energy range of-3.31 to -0.99 eV for the FeN5 system with adsorbed CO.
Zhu prepared a Fe-N4single site catalyst by the pyrolysis of the Fe-doped ZIF-8. The Fe-N4configuration was metalloporphyrin-like and highly dispersed within the nanostructures in high loading. Because of the formation of plentiful large mesopores into the nanoframes during pyrolysis,the loading of active single sites was enhanced as well as mass and charge transports. Due to these improvements, this Fe-N4catalyst displayed unique electrocatalytic performance for electrochemical CO2reduction. Interestingly, Fe-N4showed high stability with a slight decay of its selectivity during the continuous electrolysis for 12 h at the potential of -0.47 V69.
Recently, Hu and coworkers synthesized a robust single Fe site catalyst through the pyrolysis of Fe-doped zinc (Zn) 2-methylimidazolate framework (ZIF-8) under N2at 900 °C. The Fe center was highly dispersed on the surface of the in situ formed N-doped carbon (N-C) with the loading of 2.6% (w).HAADF-STEM and operando X-ray absorption spectroscopy revealed the formation of the Fe3+atoms are isolated and coordinated with pyrrolic nitrogen species of N-C (Fig. 11a,b).The N 1s XPS spectra disclosed more detailed structure of the Fe SSCs (Fig. 11c). The resulting catalysts (Fe3+-N-C) displayed an excellent electro-catalytic conversion of CO2to CO with the Faradaic efficiency (FE) of CO higher than 80% between -0.2 and -0.5 V versus RHE (Fig. 11d). During electrocatalysis, Fe3+-N-C maintained their +3 oxidation state probably through electronic coupling to the conductive carbon support which is different with its Fe2+counterpart. The superior catalytic performance was derived from the faster CO2adsorption and weaker CO absorption with Fe3+single sites37.
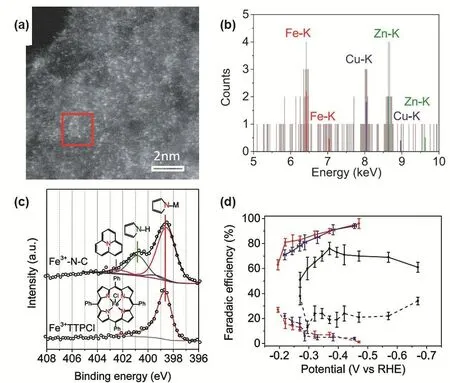
Fig. 11 (a) Aberration-corrected HAADF-STEM image, (b) EDS spectrum of the red square region. (c) N1s XPS spectra of Fe3+-N-C and Fe3+TPPCl. Red, green and pink peaks are assigned to pyrrolic N coordinated to metals, uncoordinated pyrrolic N and graphitic N,respectively, Color online. (d) Faradaic efficiency (FE) of CO (solid lines) and H2 (dashed lines) production.
Deng and coworkers reported a strategy of the preparation of Fe-SSCs by a multiscale structural modulation from macro-scale to atomic-scale. Using this method, the Fe-SSCs was successfully confined on the carbon foam and displayed enhanced catalytic performance of electroreduction of CO2to CO with FE of 94.9% at -0.5 V (vs RHE). The Fe-SSCs prepared by this method possesses pore-enriched environment, efficient electrical conductivity and abundant active sites. More importantly, the excellent performance was retained after a longterm test. The high stability was presumed to be derived from the coordination of single iron atoms with four nitrogen atom in the carbon matrix70.
To understand the correlation of reactivity and structure of the automatically dispersed M-N4(M = Fe and Co) sites for the CO2reduction reaction. Li and coworkers prepared Fe-N4and Co-N4catalysts by the pyrolysis of Fe- or Co-doped metal-organic framework precursors (Fig. 12a). The nitrogen coordinated Fe or Co sites are atomically dispersed on N-doped carbon (M-N-C).The catalytic measurements showed that Fe-N4sites are intrinsically more active than Co-N4sites in M-N-C catalysts for CO2reduction to CO with much higher FE. Computational study revealed that the edge hosted M-N2+2-C8sites which connected two armchair-like graphitic layers was able to be the active sites for the CO2electroreduction (Fig. 12b). This result is different to the reported discovery where the M-N4-C10sites was considered to be the active moieties. Interestingly, the M centers and C atoms to N atoms acted as the active sites to adsorb *CO and *OH, respectively, benefiting the C-O cleavage during the CO2reduction reaction (Fig. 12c)71.
2.1.5 Other catalysts
Apart from Fe, Co, Ni, other isolated metals such as Zn, Sb,Bi and Pd have also been reported for the electronic CO2reduction reactions. For example, Xu reported a nitrogencoordinated single Zn site catalyst for electronic CO2conversion to CO with high catalytic performance for Faradaic efficiency(FE) of 95% at -0.43 V. Combined the experimental results and DFT calculations, the four-nitrogen-anchored Zn single atom(Zn-N4) was disclosed as the main active site for CO2electroreduction. Moreover, DFT results suggested the synergistic effect between Zn and N can enhance the catalytic performance significantly by decreasing the free energy barrier of the formation of *COOH intermediate72.
Sun and coworkers fabricated single Sb site catalyst with high weight loading by simply pyrolysis the mixture of SbCl3and urea on active carbon black under inert atmosphere. In contrast to bulk Sb, Sb2O3, and Sb nanoparticles, the atomic Sb sites supported on N-doped carbon exhibited excellent electrochemical CO2reduction to CO with high selectivity with an impressive high TOF of 16600 h-1at an over-potential of-0.79 V. The remarkable catalytic performance was ascribed to effective inhabitation of proton reduction by the single Sb site catalyst73.
Recently, Li and coworkers demonstrated the preparation of Bi-SSCs and its successful application in the electrocatalytic reduction reaction of CO2to CO. The Bi-SSCs was synthesized by the thermal decomposition of a bismuth-based metal-organic framework (Bi-MOF) and dicyandiamide (DCD) (Fig. 13a).Systematic characterization demonstrated the coordination of the Bi centers with four nitrogen species nearby. The Bi-N4sites on porous carbon networks exhibited high electroreduction of CO2to CO with a high FE of up to 97% and high TOF of 5535 h-1at a low overpotential of -0.39 V (vs RHE) (Fig. 13b). In the presence of single Bi-N4site, both CO2activation and the formation of key intermediate COOH* presented low free energy barriers leading to the excellent catalytic performance(Fig. 13c)74.

Fig. 12 (a) Schematic illustration of the synthesis of M-N-C catalysts. (b) Atomic structure of M-N2+2-C8 (M = Fe or Co) active sites.(c) Calculated free energy evolution of CO2 reduction to CO on M-N2+2-C8 sites under an applied electrode potential (U) of 0 V and -0.6 V.
Although single Pd site catalysts have been widely used in lots of redox reactions, its application in CO2electroreduction is rare. Chen and coworkers reported for the first time a Pd-N4site for CO2reduction reaction. The Pd SSC was synthesized by the pyrolysis of the mixture of glucose and dicyandiamide and Pdcontaining salt. The as-synthesized Pd-N4sites showed FECOof 55% at -0.5 V (vs RHE), which was 2.5 times higher than that(22%) obtained by Pd/C at the same condition75.
Later on, a Mo SSCs was successfully fabricated where the single Mo sites was loaded on the N-doped graphene electrode and exhibited outstanding catalytic activity and selectivity on the electrocatalytic CO2reduction toward the formation of formate(Fig. 14a,b). At lower over-potential, the single Mo sites provided higher current density. Because of the high catalytic performance of Mo SSCs, improved C1product selectivity and formate production rate of 747 mmol·g-1·h-1were achieved in the presence of 4% (molar fraction) ionic liquid, far exceeding than that of N-doped graphene (Fig. 14c,d)76.
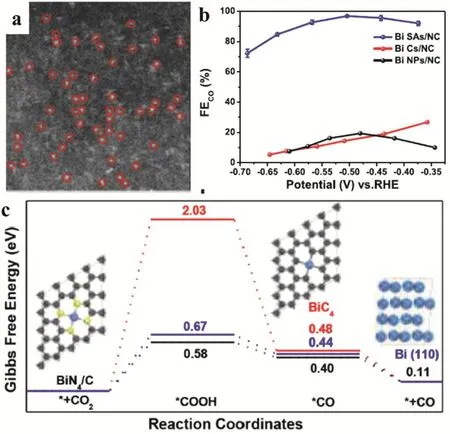
Fig. 13 (a) Magnified HAADF-STEM images. (b) FECO of CO2 electronic reduction. (c) Calculated Gibbs free energy diagrams for CO2 electroreduction to CO on different catalysts.
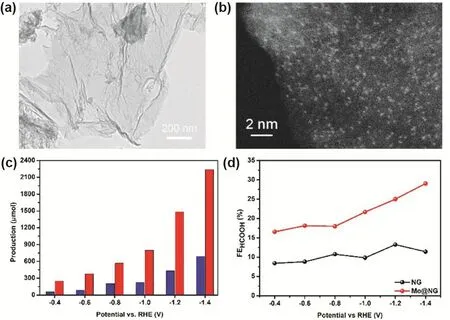
Fig. 14 (a) TEM image of as-prepared Mo@NG material. (b) HAADF-STEM image of the edge area of Mo@NG in (a).(c) The formate production comparison of NG (blue filled solid column) and Mo@NG (red filled solid column) at different potential vs RHE. Color online. (d) The formate faraday efficiency comparison of NG and Mo@NG.
2.2 C2+ chemicals synthesis
Recently, Mougel and coworkers synthesized a single Cu site catalyst by pyrolysis of the Cu coordinated ZIF-8 material (Fig.15a). The prepared sample (Cu-N-C) was successfully applied on the CO2electronic reduction. More interestingly, C2+products(ethanol and ethylene) were achieved as the major product with a Faradaic yield of 80% under 0.1 mol·L-1of CsHCO3solution at a potential of -1.2 V vs RHE (Fig. 15c,d) and CO was obtained as minor product. After the pyrolysis, the copper atoms were considered to be coordinated with four nitrogen atom forming the CuN4sites (Fig. 15b). However, the isolated CuN4sites were quickly converted into Cu NPs revealed by XAS analysis during the electroreduction of CO2. Remarkably, the single Cu site catalysts were recovered after the reaction indicating this process is reversible77.
3 Catalytic photoreduction of CO2
Solar-driven photocatalytic CO2reduction reaction (CO2RR)where the photo-catalysts are activated by the light leading to electron-hole pairs. These electron-hole pairs could be separated and act as the active sites for the CO2reduction44,78,79. Thus photocatalytic reduction of CO2is significantly attractive not only diminish CO2emission but also save energy and yield value-added chemicals. In past decades, many efforts have been devoted to developing novel catalysts that can enhance the photoreduction of CO280-82. For this respect, numerous nanomaterials such as Ru, Pt, Pd, Ni, Co and Fe, were reported and their catalytic activities can be carefully controlled by tuning the morphologies, particles sizes, compositions. Although these achievements, the limited fraction of surface active sites in these nanometer photocatalysts leads to poor catalytic performance.To maximize the utilization of the active sites, the emerging single site catalysts provide a great opportunity because of almost all metal sites are exposed on the surface, probably achieving highly efficient photocatalytic reduction of CO2. In this part, we will discuss recent advances on the photoconversion of CO2to CO, CH4and CH3OH, etc83,84.
3.1 Cobalt catalysts
Ye and coworkers prepared a Co SSCs by the incorporation of coordinatively unsaturated single Co atoms in a MOF matrix(MOF-525, Fig. 16a). Compared with the catalysts coordinated with Zn or prepared by other ligand linkage, the as-synthesized MOF-525-Co catalyst displayed excellent catalytic performance on the selectively capture and photoreduction of CO2to CO or CH4under visible-light irradiation as shown in Fig. 16b,c.Moreover, compared with MOF-525, MOF-525-Co produced a 3.13-fold and 5.93-fold improvements in CO and CH4production respectively, confirming that the catalytic performance was significantly enhanced by the introduction of Co sites. Mechanistic investigation revealed the electron-hole was efficiently separated in the presence of single Co sites leading to high selectivity. The coordination of isolated Co sites with the porphyrin units producing the active sites and can accessed to the molecular CO2and inhibited the aggregation of Co single sites85.
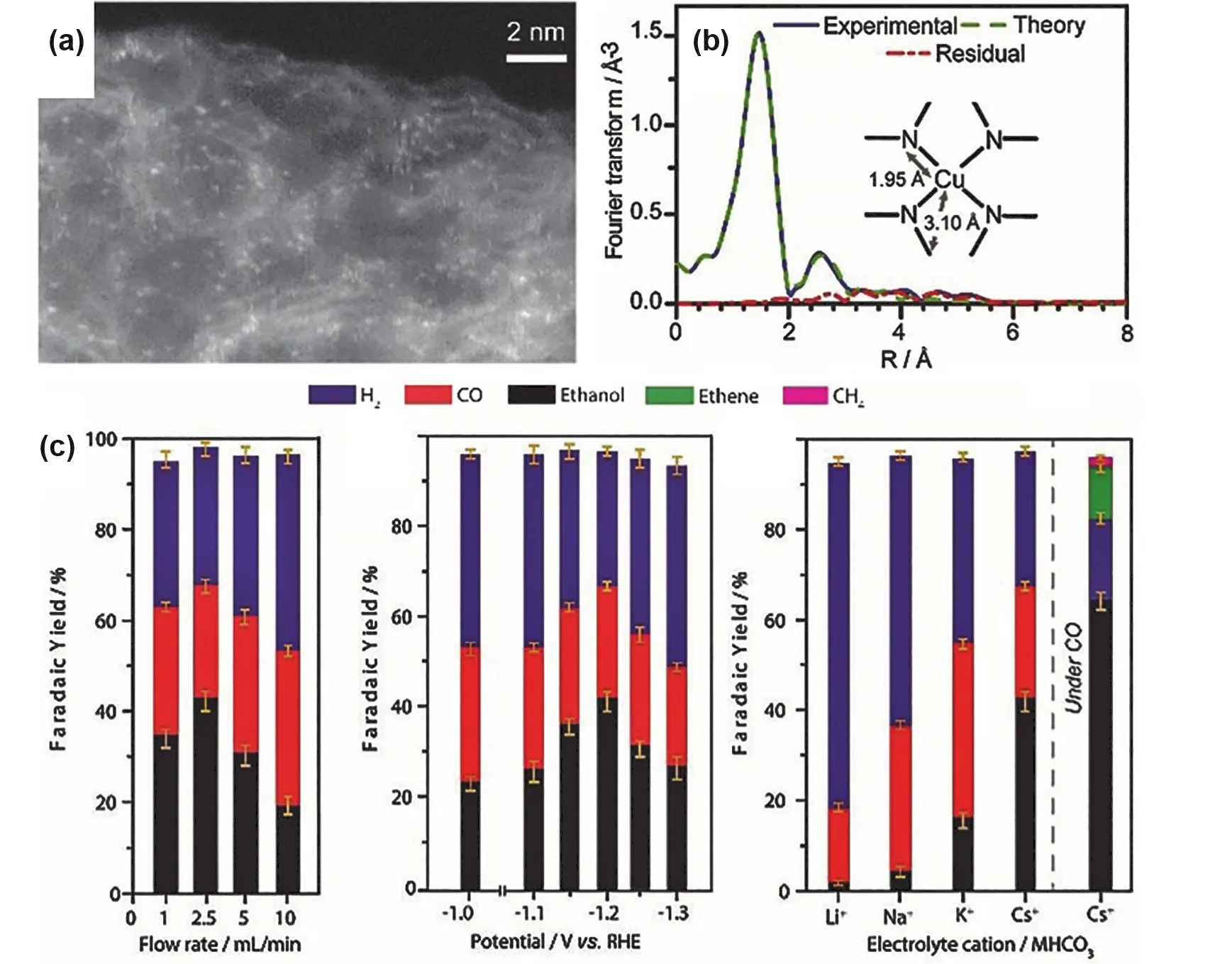
Fig. 15 (a) HAADF-STEM image, (b) Cu K-edge EXAFS analysis in the Fourier-transformed space; Faradaic yields of CO2 reduction on Cu0.5NC. Faradaic yields of (c) at @1.2 V vs RHE in a 0.1 mol·L-1 CsHCO3 aqueous solution under various flow rates of CO2 and (d) at 2.5 mL·min-1 CO2 flow-rate in 0.1 mol·L-1 CsHCO3 aqueous solution, at various applied potentials during CPE.
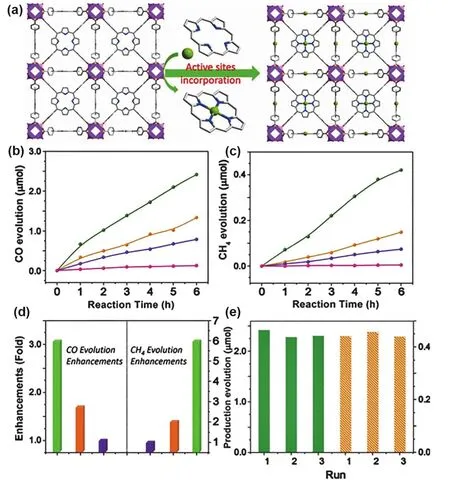
Fig. 16 (a) View of the 3D network of MOF-525-Co featuring a highly porous framework and incorporated active sites. Time dependent(b) CO and (c) CH4 evolution over MOF-525-Co (green), MOF-525-Zn (orange), MOF-525 (purple) photocatalysts, and H6TCPP ligand (pink). (d)Enhancement of production evolution over MOF-525-Co (green), MOF-525-Zn (orange), and MOF-525 (purple). (e) Production yield of CO (green) and CH4 (orange) over MOF-525-Co photocatalyst as a measure of reproducibility by cycling.
In 2018, Xiong and coworkers fabricated a single Co site catalyst by anchoring the individual single Co atoms on the partially oxidized graphene nanosheets via a thermal treatment strategy (Fig. 17a,b). Compared with the precursors, the resulting Co1-GO acted as an efficient heterogeneous catalyst for the photo-conversion CO2to CO with a high TON value of 678 for CO production and an exceptional TOF of 3.77 min-1(Fig.17c). Moreover, the Co1-GO displayed excellent stability and can be recycled for five runs and 15 h with ≈ 88% of activity retained (Fig. 17d). The partially oxidized graphene was believed to play the crucial dual roles on both providing the C/O functional sites to immobilize the isolated Co atoms and also offering relatively high conductivity to favor the transfer of photo-irradiated electrons86.

Fig. 17 (a) Schematic illustration for the synthetic procedure of the Co1-G catalyst. (b) HAADF-STEM images of the Co1-G catalyst.The atomically dispersed Co atoms are highlighted by the yellow circles. (c) TONs of CO and H2 production by Co1-G nanosheets in the first 3 h under visible-light (λ > 420 nm) irradiation, in comparison with those by graphene (G), CoCl2, graphene with CoCl2 (CoCl2 + G), and graphene oxide with CoCl2 (CoCl2 + GO) under the same condition. (d) TONs of CO and H2 production in the first 3 h using Co1-G nanosheets as a catalyst in cycling tests. Each cycle takes 3 h.

Fig. 18 (a) HAADF-STEM images of Co-Bi3O4Br, (b) EDS mapping images of Co, Bi, O, and Br. (c) Photoreduction of CO2 into CO over Bi3O4Br and Co-Bi3O4Br materials. (d) Mass spectra of 13CO (m/z = 29) produced over Co-Bi3O4Br-1 in photoreduction of 13CO2.
Later on, Li and coworkers fabricated a Co SSCs by the coordination of Co2+sites ([Co-(cyclam)Cl2]Cl) with nitrogen sites on carbon nitride (C3N4) through a simple deposition method without the absence of other organic ligands. The prepared Co-SSCs showed excellent catalytic activity and product selectivity on the photoreduction of CO2toward CO formation with a TON of over 200 under visible-light irradiation.High Co loading resulted in low activity although the selectivity was similar. Experimental and spectroscopic investigations confirmed that the presence of isolated Co2+sites was essential for the high catalytic performance87.
Liu and coworker successfully incorporated the single Co sites into the Bi3O4Br atomic layers fabricating an efficient and stable photocatalysts for CO2reduction (Fig. 18a,b). The strategy for Co-Bi3O4Br preparation was sacrifice reagent free and had no extra photosensitizer used. The resulting Co-Bi3O4Br displayed excellent catalytic activity on the photo-conversion of CO2yielding CO in a rate of 107.1 μmol·g-1·h-1, affording almost 4 and 32 times higher than that of Bi3O4Br atomic layer and bulk Bi3O4Br, respectively (Fig. 18c). The excellent catalytic performance was ascribed to the cooperation of ultrathin configuration and isolated Co sites, the Co-Bi3O4Br where the single cobalt sites facilitate the charge transition, CO2adsorption and activation. With these benefits, the energy barrier for CO2activation was lowered and the COOH* intermediates was reasonable stabilized and the rate-limiting step for the transformation of intermediate COOH* to CO* can be tuned88.
In addition, the combination of two different single metal sites was also applied in the photoreduction of CO2. Zhang demonstrated that isolated Co and Ru sites ((Co/Ru)-UiO-67)were interacted with functionalized phosphorescent MOF by a two-step self-assembly process. Interestingly, the composition of syngas from CO2photo-conversion can be extensively adjusted by changing the water contents and Ru/Co ratios in(Co/Ru)-UiO-67. When tuned the ratio of Co/Ru ratio to 2.4, the proportion of mixed gas with H2: CO = 2 : 1 was achieved by the photoreduction of CO2which is the same as used in the Fischer-Tropsch process. Under optimal reaction conditions, the syngas was obtained in high yield of 13600 μmol·g-1after 16 h,displaying much higher catalytic activity than its homogeneous counterpart89.
3.2 Copper catalysts
Ozin prepared a single Cu sites catalyst by an in situ photodeposition method with different amounts of Cu(NO3)2·3H2O in deionized water containing TiO2. The prepared Cu/TiO2exhibited excellent catalytic activity on the photoreduction of CO2to CO. The loading of the isolated Cu sites was vital for the efficient conversion of CO2. After optimization, the Cu/TiO2-2 where the content of Cu was 2% (w)displayed the highest activity of CO2photoconversion irradiating by the solar light for 2 h affording 10-fold higher of CO production than that of the pristine TiO2. Using this abundant, low-cost, nontoxic, atomically dispersed Cu photocatalyst, the photoreduction of CO2could be recycled for several time90.
In 2019, Xu and coworkers demonstrated that atomically dispersed Cu species supported on mesoporous TiO2(mTiO2)exhibited high catalytic performance on the gas-phase photocatalytic CO2reduction with H2O (Fig. 19c,d). The manufacture of Cu species grafted on mTiO2is illustrated (Fig.19a,b), where the mTiO2support had smooth surfaces and an amorphous structure. The Cu-mTiO2showed superior catalytic performance compared with TiO2and m-TiO2(Fig. 19e).
Mechanistic investigation revealed that atomically Cu(II) was initially dispersed on the surface of mTiO2and in situ reduced to Cu(I) and ultimately to Cu(0). The Cu(I)/Cu(0) mixed sites were believed to be the active centers and proposed to be more effective for CH4production from photoreduction of CO2.Furthermore, the structural properties of mTiO2vide supra benefited the CO2adsorption and charge carrier transfer, leading to the high catalytic activity91.
3.3 Nickel catalysts
Recently, Zou fabricated 2,2'-bipyridine-based COF containing a single Ni sites (Ni-TpBpy, Fig. 20a). The Ni-TpBpy displayed a synergistic effect and exhibited excellent selectivity on the photoreduction of CO2to CO in aqueous media. CO production achieved 4057 μmol·g-1after 5 h reaction with 96% selectivity (Fig. 20b). After 3 times cycles, the photocatalytic CO evolution rate could reach to 966 μmol·g-1·h-1over 3 times in a total of 6 h irradiation time, although a slight loss of activity for CO production in the third reaction observed (Fig. 20c).Different linkage units were studied and showed that Ni-TpBpy afforded the highest catalytic performance suggesting the importance of the bpy units in catalysis. The micro-coordination environment between the single Ni sites and TpBpy can stabilize the catalytic active Ni center. Moreover, the hydrogen-bond interaction in the catalyst unites can facilitate the reduction of CO2prior rather than H2evolution. These synergistic effects contributed to high catalytic activity and selectivity92.
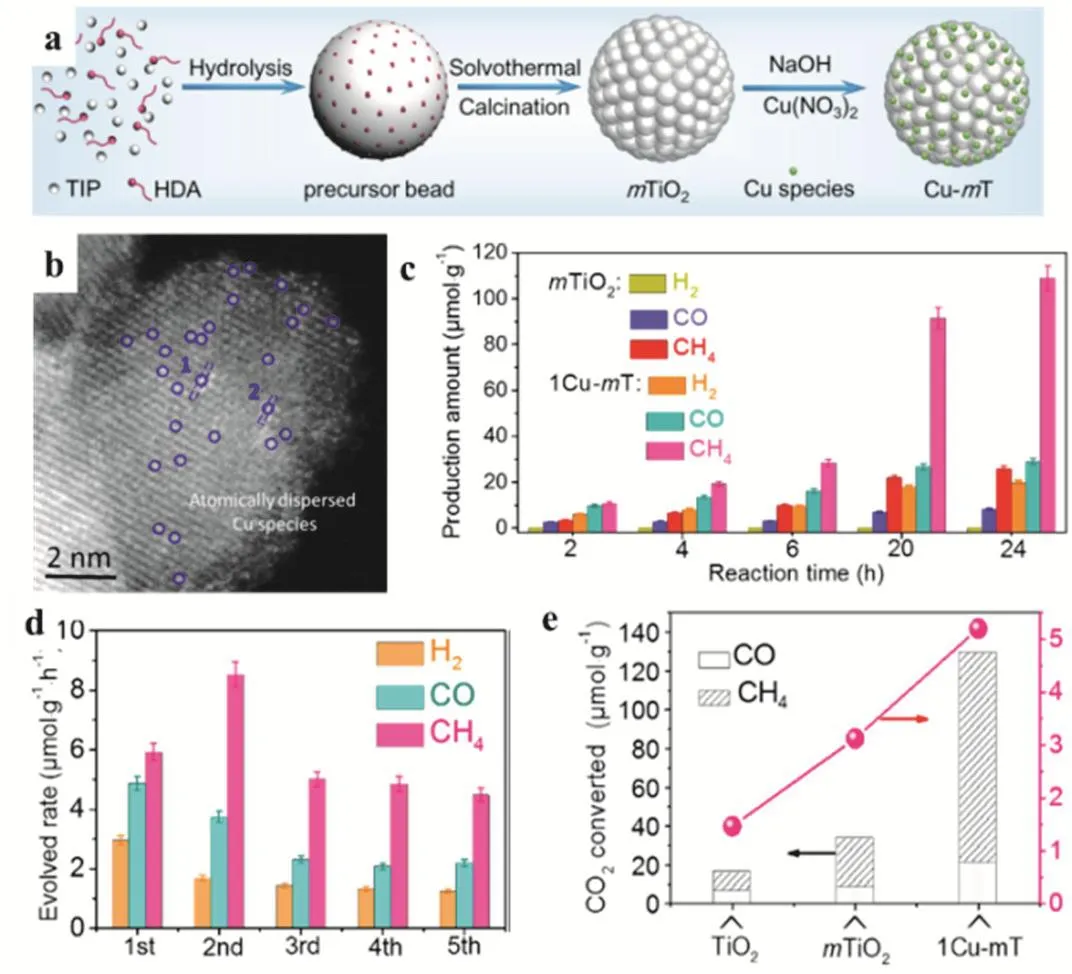
Fig. 19 (a) Illustration for the synthesis of mTiO2 and Cu-mT (mTiO2). (b) Magnified HAADF-STEM image of 1Cu-mT.(c) Time-dependent product yield over mTiO2 and 1Cu-mT. (d) Recycling experiments over 1Cu-mT during every 2 h of reaction time.(e) Converted amount of CO2 as well as produced CH4/CO molar ratios over samples of TiO2, mTiO2, and 1Cu-mT.
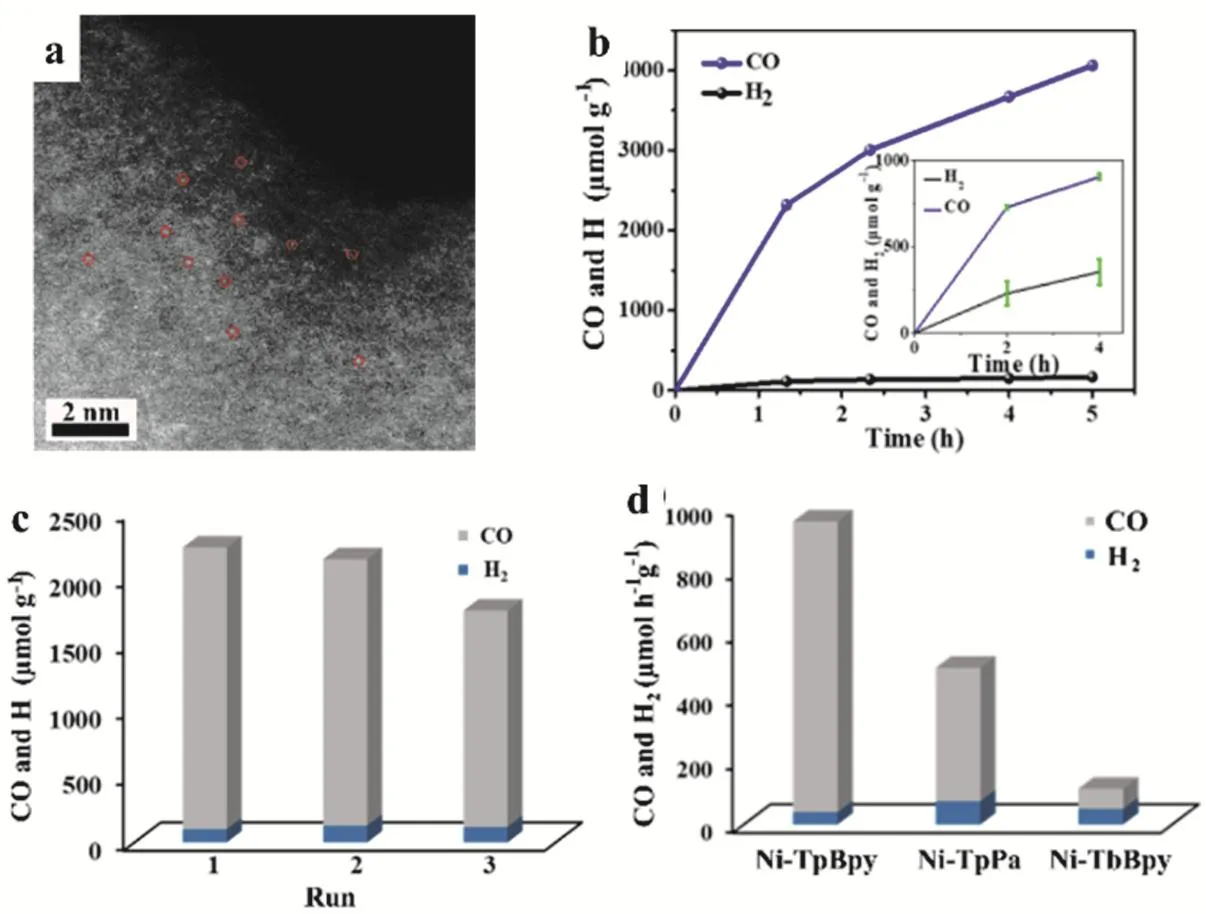
Fig. 20 (a) HAADF-STEM images of Ni-TpBpy. (b) Photocatalytic evolutions of CO and H2 by Ni-TpBpy under 1 and 0.1 atm (1 atm = 101325 Pa)(diluted with Ar, inset). (c) Stability tests of Ni-TpBpy for selective photoreduction of CO2. (d) Ni-TpBpy, Ni-TpPa, and Ni-TbBpy for photocatalytic reduction of CO2 in a 2 h reaction.
3.4 Other catalysts
Very recently, rare-earth single erbium (Er) atoms supported on carbon nitride nanotubes (Er/CN-NT) was synthesized by Li.N-doped nitride nanotubes were prepared using the melamine sponge, Er(NO3)3·5H2O and urea as precursors. The Er/CN-NT exhibited outstanding photocatalytic CO2reduction. Under the same conditions, the CO was superior formed with a production rate of 12.75 μmol·g-1·h-1while the CH4evolution rate was 0.45 μmol·g-1·h-1. The importance of single Er sites was revealed by both the experimental results and density functional theory calculations93.
Besides the non-noble single metal catalyst vide supra, the single Au site catalysts were also successfully utilized on the photoreduction of CO2. Wang synthesize a single Au metal catalyst by immobilizing Au sites on zirconium porphyrinic metal-organic framework (MOF) hollow nanotubes (HNTMs)(HNTM-Au-SA, Fig. 21a,b). HNTM-Au-SA displayed excellent photo-coupled electrocatalysis activity on the CO2reduction. Motivated by light irradiation, HNTM-Au-SA exhibited the FECOof 95.2% at 0.8 V under visible light while a maximum of FECOof 94.2% was detected at 0.9 V without light (Fig. 21d). Moreover, HNTM-Au-SA displayed a maximum TOF of 37069 h-1at -1.1 V without light irradiation.After motivated by light, a positive shift about 130 mV was observed although a similar TOF curve of HNTM-Au-SA achieved, indicating visible light can reduce the overpotential for CO2activation (Fig. 21e)94.
4 Catalytic thermal-reduction of CO2
Compared with CO2reduction conducted by electrocatalysis and photocatalysis, thermal reduction receives significant attentions because of its potential application in large scale in industry80,95,96. Moreover, in thermal catalysis, flexible combination of other active components can make the CO2conversion more favorable and effective23,24. Thus, many developments have been achieved on thermal reduction of CO2to value added chemicals3,22,25,97-99. However, most of the efficient catalytic systems are related to nanocatalysts. Although high activity and selectivity are achieved by these catalysts, only small portion of the metal sites on the surface was involved in the catalytic process lowering the efficiency of these metals. By introducing single site catalysts on the CO2reduction, higher TON and TOF could be achieved because of almost 100% utilization of the active sites. In this part, we will summarize recent advances in CO2thermal reduction by single site catalysts.
4.1 C1 chemicals synthesis
4.1.1 Platinum catalysts
Amal and coworkers prepared single Pt atoms and Pt nanoclusters anchoring on the surface of CeO2through the wet impregnation strategy (Fig. 22a). Compared with Pt nanoclusters (2Pt/CeO2), the synthesized Pt-SSC (0.05Pt/CeO2)displayed approximately 7 times higher activity on CO2reduction toward CO and CH4formation because of the atomic dispersion of Pt single atoms (Fig. 22b). Further studies demonstrated that 2Pt/CeO2and 0.05Pt/CeO2showed different reaction paths respectively. Using the isolated Pt atoms, the binding of CO with Pt centers became weak which favored the desorption of CO and inhibited both its further hydrogenation and CO poisoning leading to 100% CO selectivity and excellent catalytic stability100.
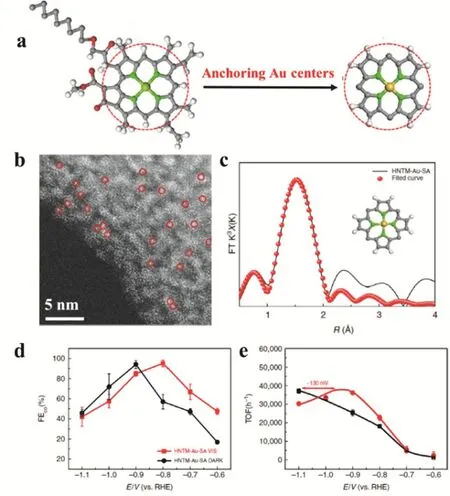
Fig. 21 (a) Preparation and nanostructure characterization of HNTM-Au-SA. (b) HAADF STEM image. Single Au atoms are highlighted in red circles. (c) The FT-EXAFS space-fitting curve of HNTM-Au-SA. Inset shows schematic models of HNTM-Au-SA, Au (yellow), N (green),C (gray), and H (white). (d) FECO on HNTM-Au-SA at the potentials of -0.6 V to -1.1 V under visible light (red line)/dark (black line).(e) TOF curves of HNTM-Au-SA under visible light (red line)/dark (black line). Error bars are ± sd 94.
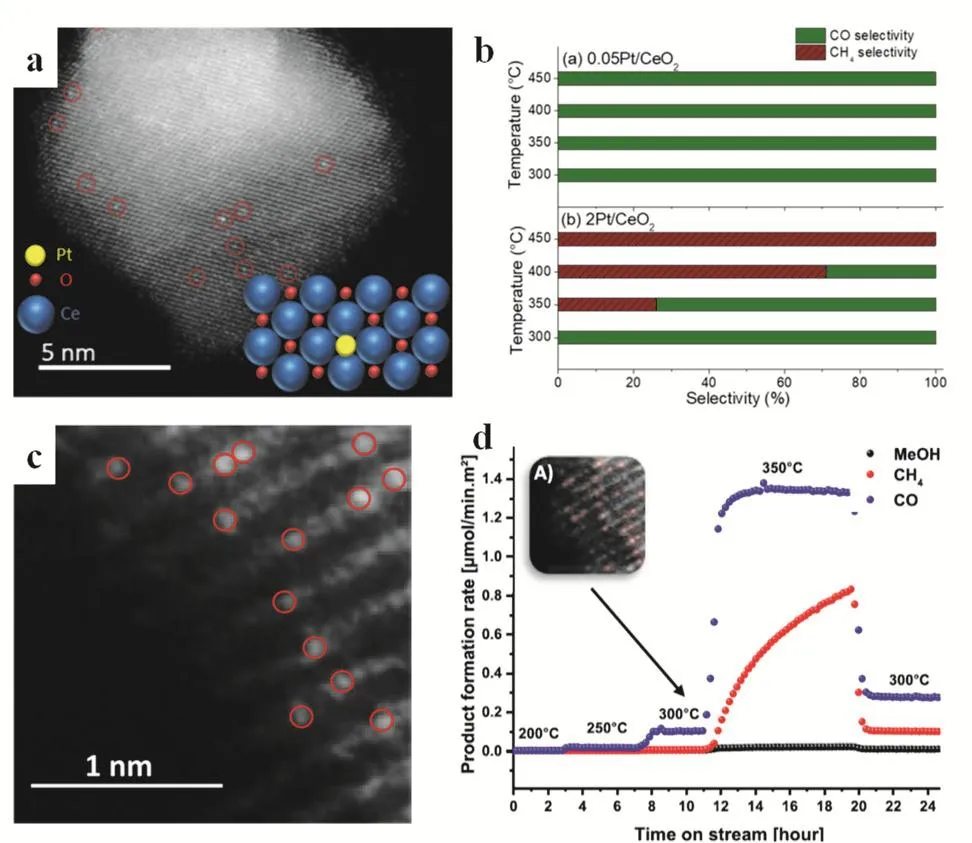
Fig. 22 (a) HAADF-STEM images of 0.05Pt/CeO2; (b) Catalytic performance selectivity of (a) 0.05Pt/CeO2 and (b) 2Pt/CeO2 toward CO and CH4 100 , Adapted from Ref. 100, Copyright 2018, American Chemical Society; (c) STEM-HAADF image of fresh Ni-SSCs, the brighter spots circled in red; (d) Product formation rate of Ni-SSCs at 30 bar (1 bar = 100 kPa), different temperatures, upper corner left represent the sample after 300 °C testing 104. Adapted from Ref. 104, Copyright 2019, American Chemical Society.
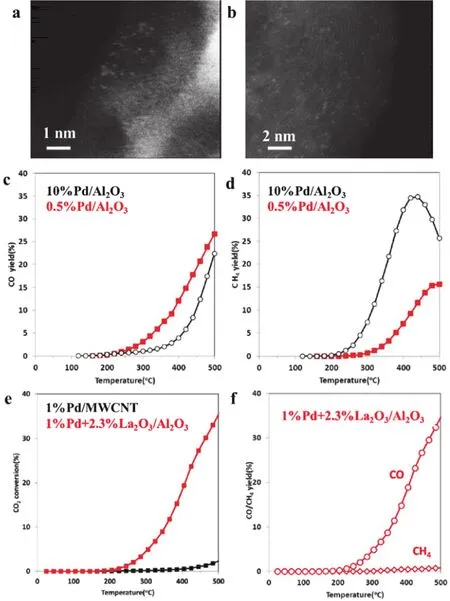
Fig. 23 (a) STEM image of 0.5% Pd/Al2O3. (b) STEM image of 1% Pd + 2.3% La2O3/MWCNT. (c) CO and (d) CH4 yield profiles during CO2 reduction reaction on 10% (black) and 0.5% (red) Pd/Al2O3. (e) CO2 conversion on 1% Pd/MWCNT (black) and 1% Pd + 2.3% La2O3/MWCNT (red), and (b) CO/CH4 yield profiles 1% Pd + 2.3% La2O3/MWCNT 101.
4.1.2 Palladium catalysts
To address the role of the atomically dispersed metal centers on oxides supports in the catalytic performances, Szanyi prepared Pd SSCs using Al2O3and La2O3modified MWCNT as supports. The Pd atoms were automatically dispersed on the surface of these supports (Fig. 23a,b). In the presence of Pd/Al2O3, the hydrogenation of CO2was carried out. CO was superior to be formed in the presence of single Pd sites catalyst(0.5%Pd/Al2O) while Pd nanoparticles (10%Pd/Al2O) exhibited higher selectivity for CH4(Fig. 23c,d). Different Pd particle sizes were obtained by changing the Pd loading possibly leading to fundamental changes in the reaction mechanism. Moreover, high CO2conversion and CO selectivity were observed using 1%Pd+ 2.3%La2O3/MWCNT compared with 1%Pd/MWCNT indicating that single Pd atoms showed lower activity on the reduction of CO2with H2in the absence of an oxide (Fig. 23e,f).Here, both La2O3promoter and Al2O3support facilitated not only the stabilization of single Pd sites but also enhancement of its catalytic activity101.
In contrast to the Pd SSCs, the single Ru atoms on CeO2exhibited 100% selectivity on CH4production. However, the Ru nanoclusters exhibited highest catalytic activity on CO2reduction toward CH4formation compared with single Ru atoms and Ru nanoparticles102.
4.1.3 Iridium catalysts
The hydrogenation of CO2to formic acid was challenging and the product normally presents as formate to drive the reaction equilibrium. Recently, Zhang and coworkers reported that porous organic polymers (POPs) was an excellent candidate to prepare single atom catalysts. Here, POP was synthesized using 1,3,5-benzenetricarbonyl chloride (TMC) with 2,6-diaminopyridine (DAP) precursors. The aminopyridine (AP)functional groups was incorporated into the polymeric framework and used for the stabilization of the single Ir metal centers (Fig. 24a,b). The resulting Ir/AP-POP catalyst exhibited excellent catalytic activity on the liquid-phase hydrogenation of CO2to formate (Fig. 24c,d)103.
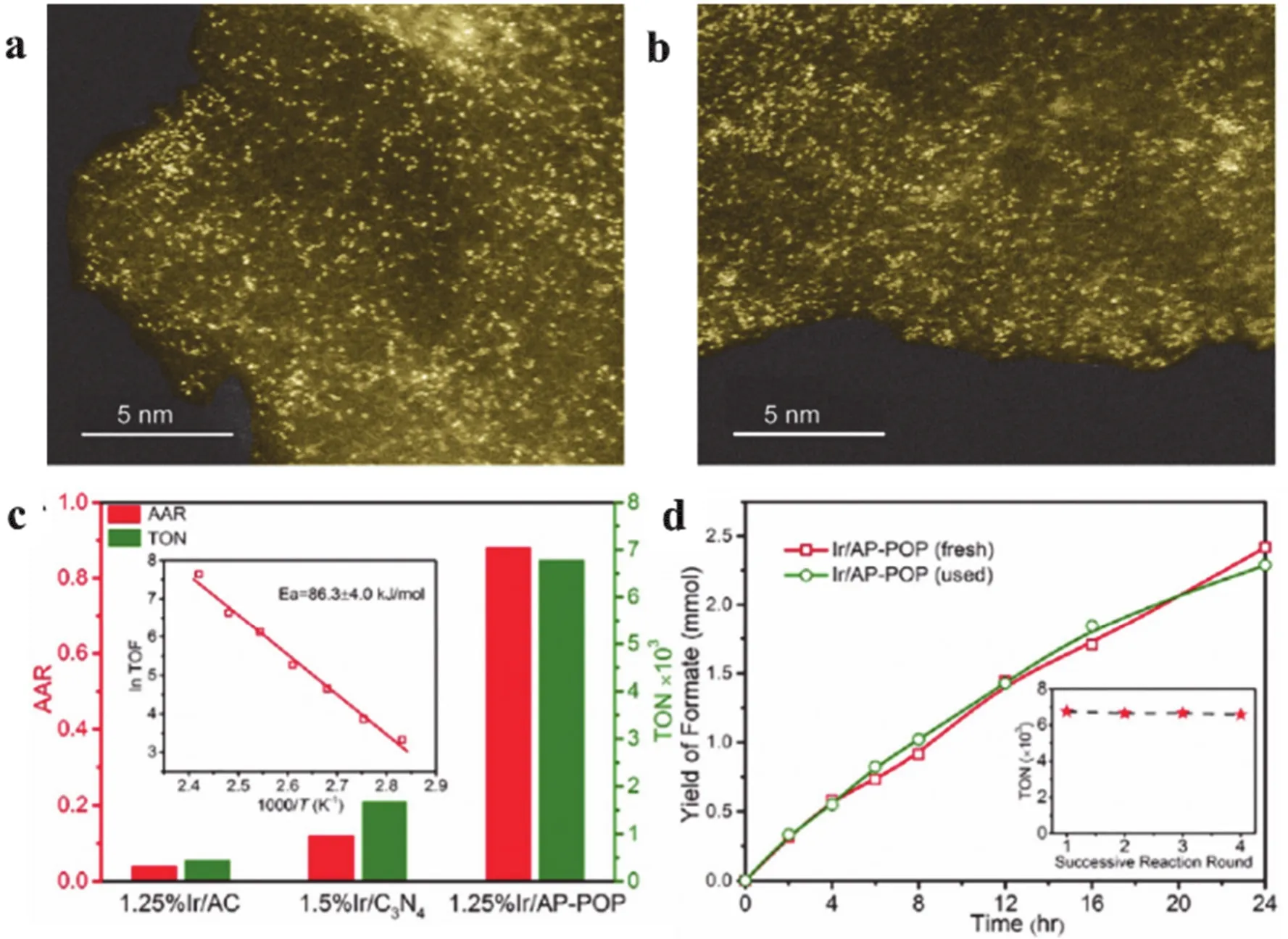
Fig. 24 (a) HAADF-STEM images of fresh 1.25% Ir/AP-POP. (b) HAADF-STEM images of used 1.25% Ir/AP-POP after CO2 hydrogenation.(c) Catalytic activities, as reflected by the formic acid to amine ratio (AAR). (d) Formate yield during the hydrogenation of CO2 over 0.66% Ir/AP-POP for the first two sequential reactions, showing essentially identical yields. Inset: reuse of the 1.25% Ir/AP-POP catalyst with no activity loss for up to four runs 103.
4.1.4 Nickel catalysts
Recently, Frei developed a strategy for the synthesis of single Ni sites in the MgO lattice by the simple wet chemistry. The NixMg1-xO catalyst can be manipulated in the range of 1%-10%(atom fraction). The structure and composition of Ni SSCs were thoroughly characterized (Fig. 22c). The NixMg1-xO catalyst was active for thermal-reduction of CO2toward to CO with excellent stability (Fig. 22d). The stable performance was ascribed to the amount of single Ni atoms on the surface of the catalysts excluding the dominant effect from the bulk catalyst. DFT and kinetic investigations exhibited that a carbonate-based overlayer was formed during CO2hydrogenation and the decomposition of carbonate appears to be helpful for the Ni-SSCs dispersion. The isolated Ni atoms suppressed the further hydrogenation of CO to CH4or MeOH which occurred normally by Ni clusters affording higher selectivity. Moreover, compared with Ni clusters, the single Ni sites are limited to catalyze a multi-electron reaction leading to the excellent catalytic performance104.
Ye and coworkers fabricated a single Ni site catalyst using two dimensional amorphous Y2O3nanosheets as support. The resulting SA Ni/Y2O3catalyst exhibited excellent catalytic activity on the thermal CO2hydrogenation with almost 100% selectivity for CH4formation (Fig. 25a,b). ~87% of conversion could be retained after 90 h hydrogenation and no obvious difference was observed on the morphology of the SA Ni/Y2O3after catalysis indicating its catalytic stability (Fig. 25c,d). The photothermal methanation of CO2using SA Ni/Y2O3nanosheets was proceeded assisted by a selective light absorber (Fig. 25e).The highest temperature of SA Ni/Y2O3could be reached up to~285 °C under 1.0 kW·m-2irradiation. Because the addition of selective light absorbers facilitated both the adsorption of full solar spectrum and production of thermal radiation. The photothermal CO2methanation can be initiated at only 0.4 kW·m-2irradiation and almost 90% of conversion was observed under 1 sun (Fig. 25f). Moreover, the addition of the selective sunlight absorber did not reduce the CH4selectivity on SA Ni/Y2O3105.
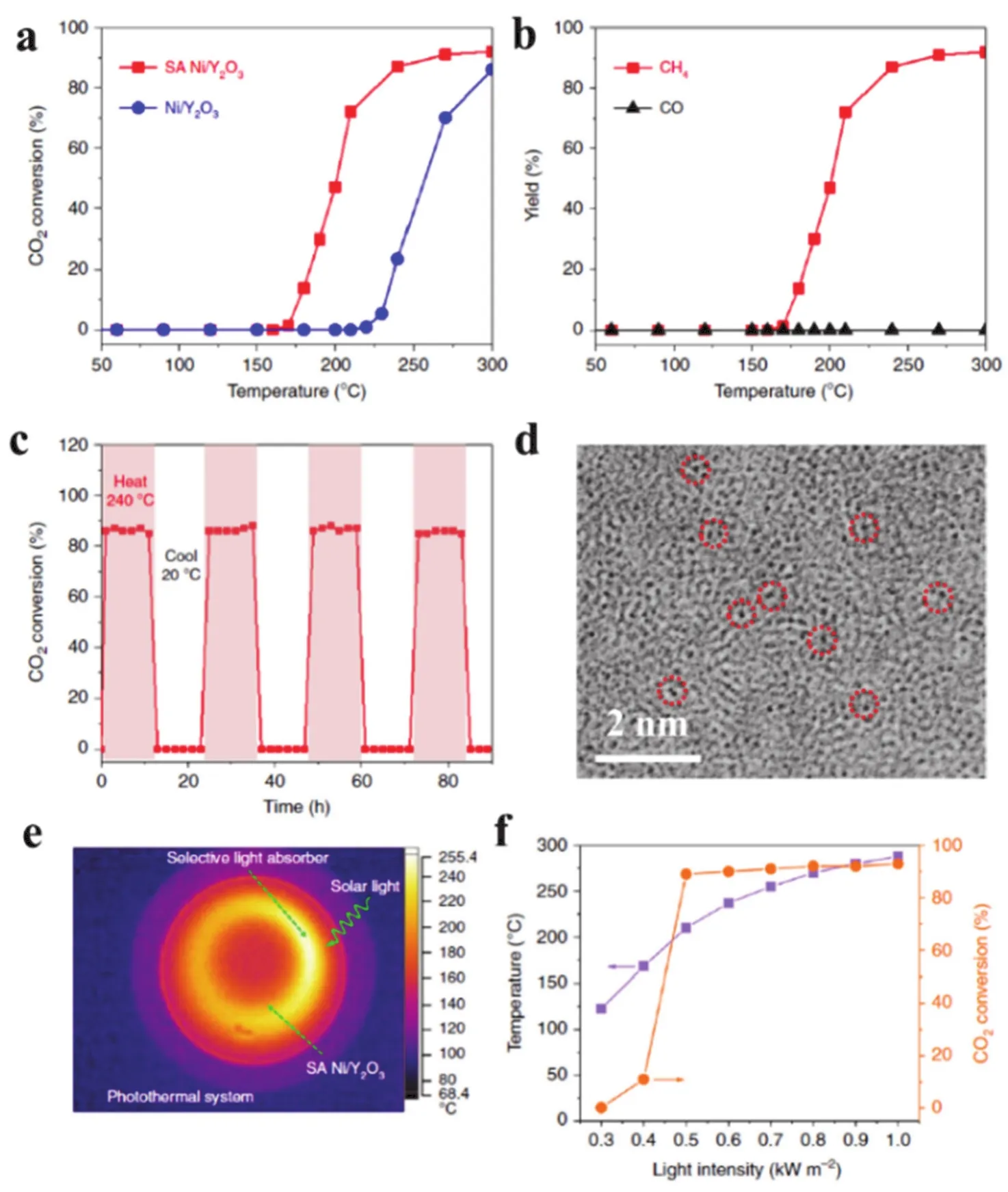
Fig. 25 (a) Thermal CO2 conversion using the SA Ni/Y2O3 nanosheets (SA Ni/Y2O3) and Ni nanoparticles/Y2O3 nanosheets (Ni/Y2O3) as a function of temperature. (b) CH4 and CO yields from the CO2 hydrogenation over the SA Ni/Y2O3 nanosheets as a function of temperature.(c) CO2 hydrogenation versus reaction time over the SA Ni/Y2O3 nanosheets at 240 °C. (d) Aberration-corrected TEM image of the SA Ni/Y2O3 nanosheets after used. (e) Spatial temperature mapping of the selective light absorber-assisted quartz tube coated with the SA Ni/Y2O3 nanosheets under 1.0 kW·m-2 of simulated solar irradiation obtained by an infrared camera. (f) The temperature and CO2 conversion achieved by the SA Ni/Y2O3 nanosheets with the selective light absorber-assisted photothermal system under different intensities of simulated solar light.
4.2 Ethanol synthesis
Llorca demonstrated the single Pd site catalyst (Pd/Fe3O4) was conducted on the hydrogenation of CO2into ethanol. At temperature range of 250-300 °C, the Pd/Fe3O4displayed stable catalytic performance and high yield was obtained with excellent selectivity toward ethanol. However, further increasing the reaction temperature, the single Pd atoms was aggregated into Pd nanoparticles leading to low activity on ethanol formation.Compared with other inorganic oxides, a specific interaction between single Pd atoms and Fe3O4occurred and afforded a specific manner for C-C coupling106.
4.3 Other chemicals synthesis
In 2019, Li and coworkers prepared a Pt SSCs where the single Pt centers was stabilized by exploiting the rich Ti-deficit defects support (Ti3-xC2TyMXene, Fig. 26a,b). With a very small amount of Pt (0.4%, molar fraction), the Pt1/Ti3-xC2TyMXen exhibited excellent catalytic activity on the N-formylation of CO2and aniline using silanes under low CO2pressure. Both the conversion and selectivity reached up to 100%. And, the TON for the desired products obtained from Pt1/Ti3-xC2TyMXen was much higher than that of commercial Pt catalysts and Pt (NPs)/Ti3-xC2Ty (Fig. 26c,d). These results were ascribed to the maximum utilization of the precious metals.Moreover, Pt1/Ti3-xC2TyMXen still displayed high catalytic performance after several runs indicating its stability. Some Tideficit defect sites was replaced by the single Pt atoms forming a strong Pt-C bonds possibly stabilizing the active Pt sites107.

Fig. 26 (a) HAADF-STEM image of Pt1/Ti3-xC2Ty. (b) Magnified HAADF image of the area in the yellow box in a. Schematic columns of atoms are overlaid on the experimental images (inset). (c) Catalytic performance of the N-formylation of aniline using different catalysts.(d) Recycling test of Pt1/Ti3-xC2Ty for the catalytic N-formylation of aniline.
Recently, Beller and coworkers immobilized single Zn sites on the N-doped carbon matrix by pyrolysis of an active-carbonsupported phenanthroline-ligated Zn(OAc)2complex. The resulting Zn-SSCs can catalyze the cycloaddition of epoxides and carbon dioxide under mild conditions. Compared with a Znbased nanoparticle (Zn-NP) catalyst, Zn-SSCs showed improved activity and stability for the cycloaddition of epoxides.Importantly, under optimal conditions, a variety of carbonates were successfully produced in high yields with broad substituted epoxides108.
5 Summary and perspective
This review presents a summary of the well-defined structure of the single sites catalysts that displays unique advantages as catalysts. With their intrinsic merits, the single sites catalysts are widely studied on the conversion of CO2to CO, methane,methanol, and C2+hydrocarbons and oxygenate by photocatalysis, electro-catalysis, and thermo-catalysis. Compared with the conventional heterogeneous catalysts that used on the CO2transformations, single sites catalysts have been exhibited extraordinary catalytic activity for electrocatalytic,photocatalytic and thermocatalytic conversion of CO2into value added chemicals due to their maximum utilization of metal sites.Furthermore, the single sites catalysts provided not only significant benefits in catalytic activity on CO2hydrogenation over conventional catalysts but also provide a new platform that allows for studying the reaction mechanism in molecular levels.For example, on the electrocatalytic CO2 reduction, the electronic properties of single sites could be tuned by lattice strain around and ligand effects on the surface functional groups allowing catalyst optimisation to enhance product selectivity and current density. Meanwhile, higher conversion of CO2and selectivity towards desired chemicals could be achieved by introducing various catalytically active sites in the same material.
Single sites catalysts also demonstrate unique catalytic performance on photoreduction of CO2due to its maximum utilization of the active site, making them well-suited to photocatalytic transformation of CO2to value added chemicals especially in CO synthesis. In photoreduction of CO2, single site structures are ideal choices to utilize the interaction between the active sites and support where the suitable modification of the surface by the coordinated organic groups could be probable to suppress charge recombination, harvest visible light, and integrate materials with high CO2 chemisorption properties. For example, the hydrogen-bond interaction between the active unites with the modified support can facilitate the reduction of CO2prior rather than H2evolution.
In the thermal reduction of CO2, single sites catalysts are also displayed superior catalytic performance on the transformation of CO2to CO and methane. More importantly, C2+chemicals could be produced by this method. Interestingly, N-formylation of amines with CO2and cyclic addition of epoxides with CO2can conducted successfully in the presence of the single site catalysts. Thanks to the maximin utilization of the active sites,high TON and TOF are achieved in these reactions.
Although these achievements discussed vide supra, the catalytic reduction of CO2by single site catalysts continues to attract numerous interests from both academic and industrial points of view. Considering all these advancements, what remains the major challenges of catalytic reduction of CO2by single site catalysts in the coming decade?
Progresses on the application of single site catalysts in the electrocatalytic, photocatalytic, and thermal catalytic conversion of CO2to value added chemicals is still at an early stage. It is highly desirable to conduct further researches both in synthesis strategies of the singles site catalysts and intrinsic understanding of reaction pathways. The comprehensive understanding of structure activity relationships in the presence of singles site catalysts would be a challenge and this will facilitate not only deep insight of the CO2reduction reaction but also future rational catalyst design that be able to efficiently convert CO2into products with high activity and selectivity. Combined with computational modeling of single sites structures, these investigations on catalytic activity and mechanism would be strongly promoted.
Single site catalysts have been well fabricated in the lab scale.However, another challenge is raising about whether these catalysts can be made cheaply and with good quality control that be able to benefit its potential applications in industry. Moreover,compare with the conventional heterogenous catalysts, can these novel single site catalysts be regenerated simply when the catalytic activity lost after reactions? Unfortunately, there are less investigations on these respects. Thus, economically competitive processes should be designed for the scale-up synthesis of single site catalysts. Furthermore, the future focus should also be on synthesis of the single site catalysts with longterm stability under realistic operating conditions especially for the thermal catalytic reduction of CO2.
Based on the discussion of recent progress in the reduction of CO2by electro-catalysis, photo-catalysis, and thermal catalysis,we note that most of the works are conducted by single approach.Although several reports are disclosed with the combination of(photo)electrochemical catalysis or (photo)thermalchemical catalysis, the studies are still at a very early stage, we proposed ideas for further improvements on the interdisciplinary research of the electro-catalysis, photo-catalysis, and thermal catalysis on the purpose of efficient and selective conversion of CO2to desired valued added chemicals.
