
石油中不同环数芳烃化合物的精细分离和制备
2021-06-02 10:38李美俊唐友军何大祥KONANGuessanFrancoisDeSales师生宝
石油实验地质 2021年3期
刘 雪,李美俊,,唐友军,何大祥,,KONAN N’Guessan Francois De Sales,师生宝,朱 雷
(1.长江大学 油气资源与勘探技术教育部重点实验室,武汉 430100;2.长江大学 资源与环境学院, 武汉 430100;3.中国石油大学(北京)油气资源与勘探国家重点实验室,北京 102249;4.中国石油大学(北京) 地球科学学院,北京 102249)
多环芳烃化合物含有苯环结构,比饱和烃具有更加稳定的化学性质,在高过成熟原油和有机质、轻质油、凝析油等可能遭受过次生改造的原油中都能稳定分布[1],因而在油气地球化学研究中具有重要的作用。由于构成多环芳烃的苯环个数、取代基的个数和位置的不同,造成数目众多的多环芳烃系列化合物共存,这对后续分子组成、同位素等信息的获取造成了明显的困难。前人研究表明,芳烃化合物能够提供重要的地球化学指标[2],但相对于饱和烃类分子标志物,多环芳烃化合物的同位素组成研究还不多,特别是单个化合物的稳定碳同位素组成相关的研究成果还很少[3-4]。前人研究成果显示,不同环数多环芳烃化合物的稳定碳同位素值存在明显差别,甚至很多烷基取代位置异构体的碳同位素值也不同,这些差异性可能蕴藏了关于其母质来源和沉积环境等重要信息[3, 5-6]。例如,芳烃单体烃碳同位素组成在热演化,油气运移,油源对比[7-9]等方面能起到特殊的指示作用[10-15],因此对芳香烃单个化合物碳同位素进行系统分析,对进一步探索该类化合物的地球化学意义,及推广其在石油勘探中的应用具有重要的作用[16]。
多环芳烃化合物单体烃碳同位素组成研究的最大难题是,在常规的气相色谱分析中化合物峰共溢出和相互干扰严重,这就给单体烃碳同位素组成的测定带来了很大的困难。为了解决这个问题,很多学者在分离前处理技术上对色谱柱填充物的种类和方法、分离试剂的极性和用量上做过一些探索性实验[17-20]。理论上中性氧化铝对芳烃组分具有吸附作用,在极性较弱的洗脱试剂中加入适量极性较强的试剂时会克服氧化铝与芳烃化合物之间的吸附作用力,从而将不同环数的多环芳烃洗脱出来[21-23]。但这种方法在不同极性试剂的种类和配比上并没有统一的标准,我们无法确定使用什么样的配比最适合自己的样品,也给芳烃单体烃碳同位素的研究带来了一定的难度。为了克服色谱同位素质谱仪(GC-C-IRMS)在检测中遇到的本底大,化合物容易共溢出,未分辨复杂混合物(UCM鼓包)等问题,需要找到一种能够精细分离不同环数多环芳烃的前处理实验方法,这样不仅会大大提高单体烃碳同位素值检测的精确性,同时也会降低化合物同分异构体识别的难度[24-25]。本文对不同环数芳烃分离方法所使用的试剂进行不同极性的配比,期望通过不同环数芳烃化合物亚组分的制备获取,为芳烃化合物单体碳同位素组成的分析提供一种更加实用和有效的方法。
1 样品与试剂
1.1 样品
选取中西非裂谷系尼日尔Termit盆地白垩系Yogou组3个不同深度的原油样品进行实验,原油样品分别来自Sokor SD-1井(深度为3 218.2~3 221.0 m)、Sokor SD-1井(深度为3 245.6~3 255.1 m)和Achigore Deep-1井(深度为3 167.9~3 173.7 m),均为黏稠、能流动的黑色重质油。Termit盆地的基本石油地质概况和油气成藏特征已有大量文献报道,油源对比结果表明,Termit盆地大部分原油来自白垩系Yogou组海相泥页岩烃源岩[26-29]。
1.2 试剂与材料
石油醚(分析纯),二氯甲烷(分析纯),柱色谱所用的硅胶(选取0.149~0.074 mm的层析硅胶,用氯仿抽提至不发荧光,烘干后在140~150 ℃电热干燥箱中活化8 h,在干燥器中冷却后装入磨口瓶中备用,使用前再活化4 h)和氧化铝(选取0.149~0.074 mm的中性层析氧化铝,在400~450 ℃马弗炉中烘烤4 h,取出稍冷,移入干燥器中冷却后装入磨口瓶中,置于干燥器中保存备用),脱脂棉(用氯仿抽提72 h),层析柱(内径9~10 mm,长度12 cm),若干小烧杯和注射器。
2 实验流程
2.1 样品去沥青质
称取60 mg左右的原油样品放入小烧杯中,加入约10 mL的石油醚,使用超声波震荡仪震荡2 min之后,再用塞有脱脂棉的漏斗过滤掉沥青质,将去掉沥青质组分的原油样品在通风橱中让溶剂自然挥发,待分离。
2.2 “一步法”分离步骤
在层析柱底部填塞适量脱脂棉,先加入2 g层析硅胶,再加入2 g中性氧化铝,轻轻敲击层析柱使固定相填充均匀,立即加入适量的石油醚润湿层析柱。当石油醚液面接近中性氧化铝固定相顶部界面时,将样品滴入层析柱中。在样品液面接近固定相顶部界面时,使用30 mL的石油醚冲洗饱和烃之后,配制石油醚/二氯甲烷(体积比,下同)为99/1、95/5、9/1、1/2的混合溶剂各6 mL进行洗脱,目的是按照芳烃不同环数将单环、双环和三环化合物分离开。最后采用二氯甲烷/甲醇(9/1)的混合溶剂10 mL收集非烃馏分。
2.3 “两步法”分离步骤
(1)第一步:在层析柱底部填塞适量脱脂棉,先加入2 g层析硅胶,再加入2 g中性氧化铝,轻轻敲击层析柱使固定相填充均匀,立即加入适量的石油醚润湿层析柱。当石油醚液面接近中性氧化铝固定相顶部界面时,将样品滴入层析柱中。在样品液面接近固定相顶部界面时,使用30 mL的石油醚冲洗饱和烃,20 mL的石油醚/二氯甲烷(1/2)的混合溶剂冲洗芳烃,10 mL的二氯甲烷/甲醇(9/1)的混合溶剂冲洗非烃馏分。分离好的各组分在通风橱内自然风干,芳烃组分待下一步分离。
(2)第二步:在干净的层析柱中填充3.5 g的氧化铝,立即用适量的石油醚润湿层析柱,将上一步中浓缩的芳烃组分滴入层析柱中进行二次分离,目的是将芳烃组分分成单环、双环和三环等各部分。配制石油醚/二氯甲烷体积比为99/1、95/5、9/1、8/2、0/100的梯度溶剂去进行洗脱。
如图1所示,原油经过一次层析柱的分离实验称为“一步法”,经过两次称为“两步法”。本文采用了“一步法”和“两步法”两种分离方法进行对比实验,在经过GC-MS检测分析比较后,总结出了适合分离不同环数芳烃化合物较为有效的方法。
2.4 色谱—质谱(GC-MS)分析条件
采用GC-MS联用仪对芳烃化合物检测,仪器型号为Agilent 6890 GC-5975i MS,配置了弹性石英毛细管色谱柱(30 m×0.25 mm×0.25 μm)进行化合物的分离,载气为He,采用恒流模式(载气流量1 mL/min);色谱炉温的起始温度为80 ℃(保持1 min),然后以5 ℃/min升至310 ℃,恒温18 min。质谱采用EI电离模式(70 eV),离子温度为230 ℃,质量扫描范围m/z50~600。
3 实验结果与讨论
将上述步骤分离出来的芳烃化合物亚组分系列进行GC-MS检测分析,分别采用烷基苯特征离子m/z91,92,105,106,119和萘系物的特征离子m/z128,142,156,170,184以及菲系物特征离子m/z178,192,206,220,234进行识别[30-31]。
3.1 “一步法”检测结果
“一步法”是在传统族组分分离的基础之上,将芳烃馏分的洗脱溶剂通常为石油醚/二氯甲烷(1/2)尝试替换为梯度溶剂,从而实现过一次层析柱就将芳烃不同环数的化合物分离开,经过GC-MS的检测,结果见表1。其中石油醚/二氯甲烷(99/1)检测到了出峰非常不明显的少量烷基苯化合物,但根据总离子流图很难辨认出来。石油醚/二氯甲烷(95/5)和(9/1)的溶剂冲洗下来的馏分中都没有检测到萘,说明萘可能在分离过程中由于含量过低挥发消失了。但都检测到了多甲基萘系物,说明石油醚/二氯甲烷(95/5)的溶剂并不能将双环芳烃冲洗干净,而石油醚/二氯甲烷(9/1)的溶剂冲洗下来的浓度是前者的10倍,说明适量增加洗脱剂的极性可以有效地分离出目标化合物。石油醚/二氯甲烷(1/2)可以将菲类化合物洗脱出来,但也包含了部分的三甲基萘和四甲基萘化合物,由图2可以清楚地观察到在保留时间大约为17~22 min之间出峰的化合物被分配到了两种不同配比的试剂中,说明此种分离方法不可以达到按照不同环数分开的分离效果。
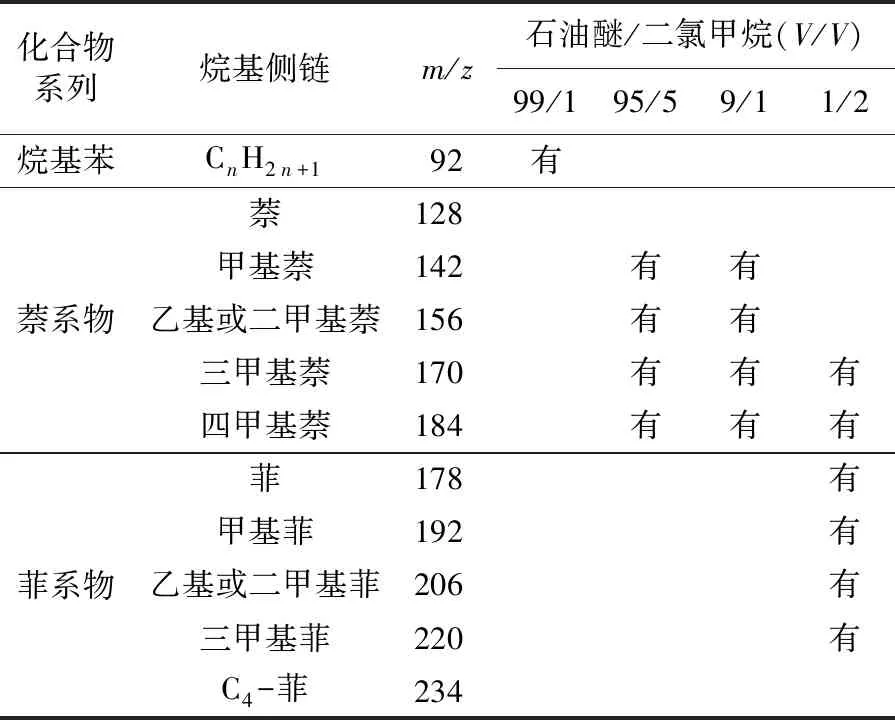
表1 Termit盆地原油样品中芳烃化合物亚组分“一步法”检测结果Table 1 Results of “one-step” separation for aromatic sub-fractions in crude oil samples from Termit Basin
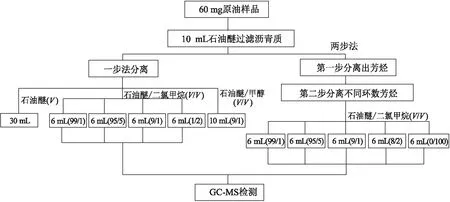
图1 “一步法”和“两步法”层析柱分离实验的分离流程Fig.1 Separating procedures of “one-step” and “two-step” methods
在柱层析实验中由于上层填充的氧化铝大多被吸附性较强的非烃化合物所占据,大多数的芳烃已经随着大量石油醚的冲洗而流动到了下面的硅胶层,硅胶对芳烃的吸附力没有氧化铝强,硅胶可以使芳烃在极性试剂的冲洗下使吸附力小的低环芳烃率先向下扩散。根据“一步法”的检测结果发现石油醚/二氯甲烷(9/1)冲洗下来的萘系物浓度是石油醚/二氯甲烷(95/5)的10倍,说明9/1的比例更适合分离双环类芳烃化合物。此外,还发现9/1与2/1洗脱出来的双环芳烃化合物和三环芳烃化合物分界不是很明显,特别是四甲基萘和芴类化合物分开得不是很彻底,而单体碳同位素的测定不仅仅需要化合物基线分离,对浓度也有很高的要求,因此,有理由认为不是仅仅使用不同极性的洗脱剂就可以分开双环芳烃系列和三环芳烃系列。
在3个原油样品分离结果中发现保留时间16.8~21.8 min是石油醚/二氯甲烷(9/1)和(1/2)两种配比试剂出峰共溢出较多的区间,说明经石油醚/二氯甲烷(9/1)溶剂冲洗之后还残留了一部分的三甲基萘和四甲基萘,其中四甲基萘在两种试剂中冲洗出来的浓度相差不大。
3.2 “两步法”检测结果
“两步法”是将芳烃馏分经过浓缩后进行的二次分离,分离结果如表2所示,这种方法在一定程度上精确地分开了烷基苯、萘系物和菲系物(单环、双环和三环芳烃)。如图3所示,石油醚/二氯甲烷(99/1)试剂分离的第一组馏分呈现规则的等间距分布,经过识别判定此类化合物为烷基苯;石油醚/二氯甲烷(9/1)试剂分离出的第三组馏分中的化合物在保留时间13~21 min出峰,经过识别判定大部分的化合物为二甲基萘、三甲基萘和四甲基萘;石油醚/二氯甲烷(8/2)的试剂分离出的第四组馏分在保留时间16~21.8 min有少量化合物出峰,经过识别判定为芴、甲基芴、二苯并呋喃和甲基二苯并呋喃,在保留时间22~28 min中识别出了菲、甲基菲和二甲基菲。
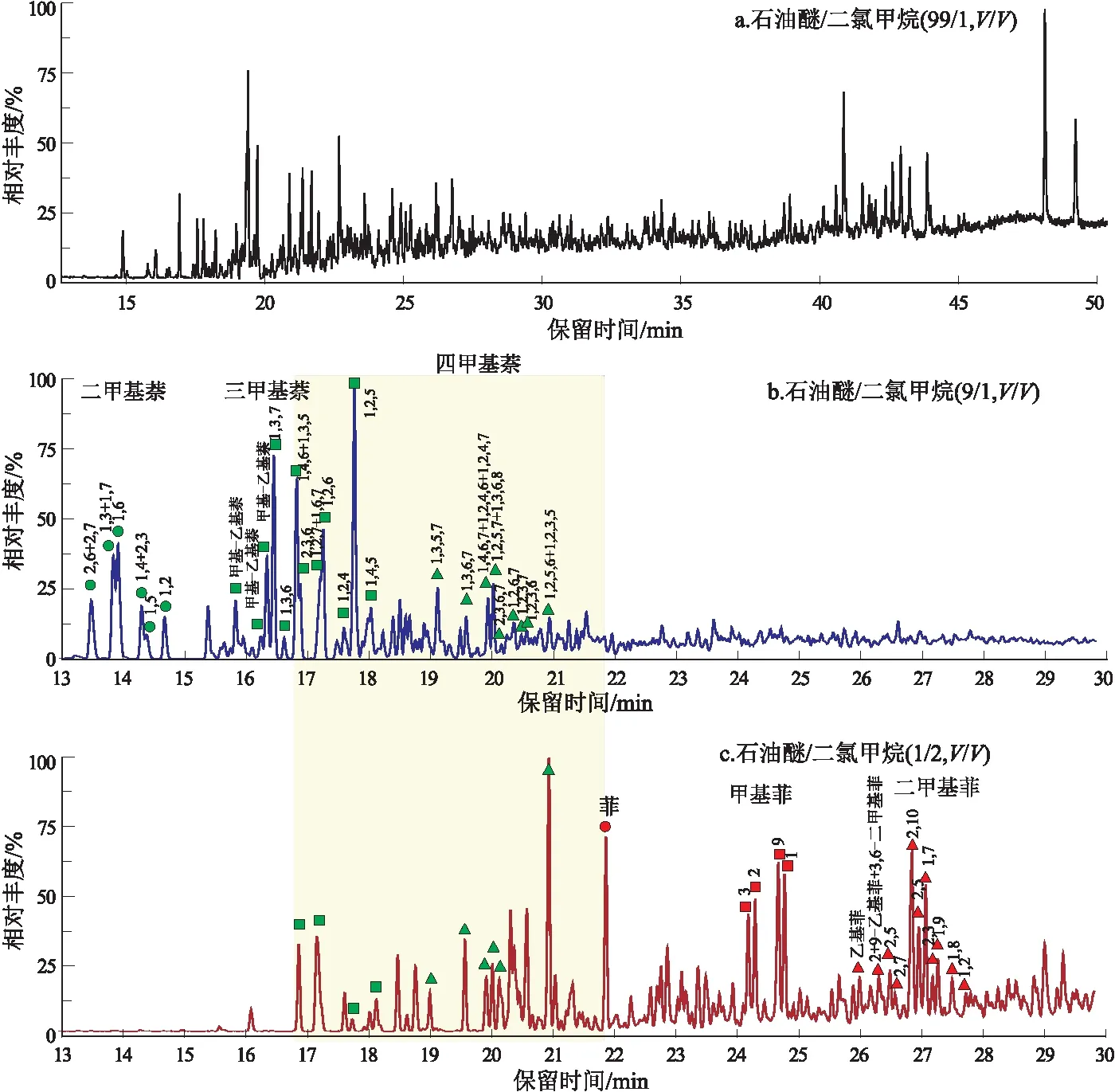
图2 Termit盆地原油样品“一步法”中使用石油醚/二氯甲烷试剂洗脱出的化合物总离子流图 峰上的数值代表取代基的位置Fig.2 Total ion flow diagram of compounds eluted with petroleum ether/dichloromethane solvents in “one-step” process of crude oil samples from Termit Basin
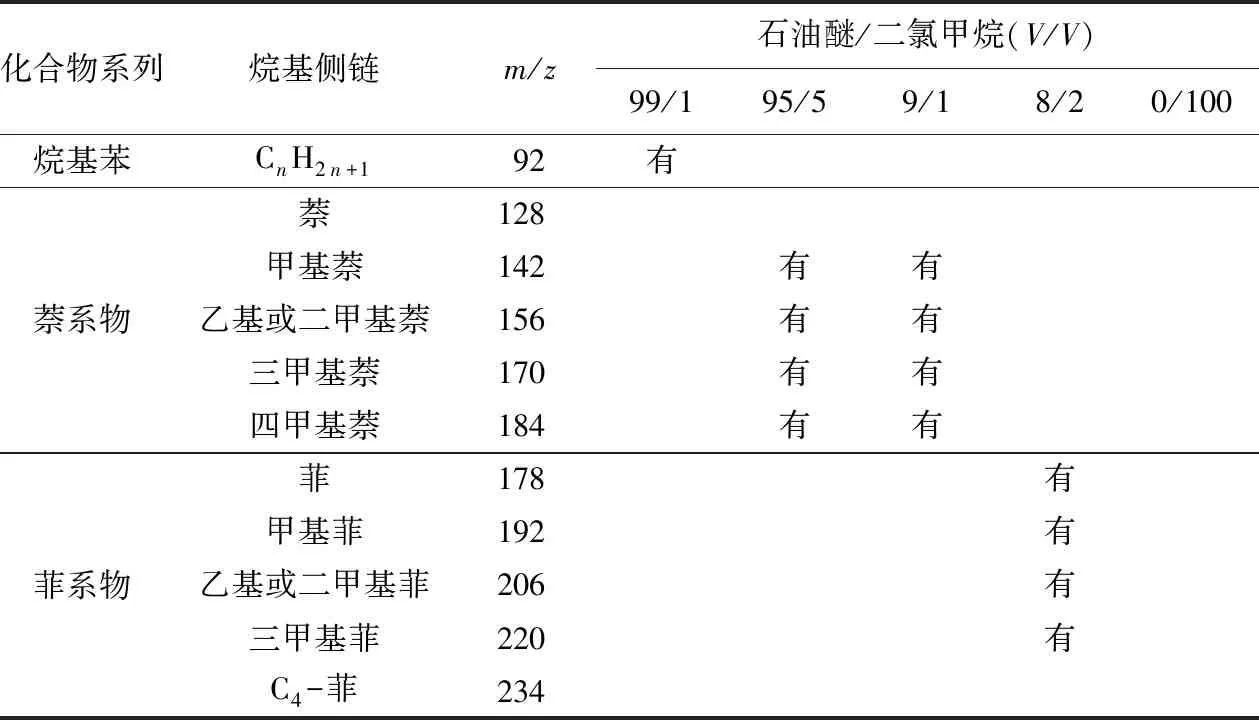
表2 Termit盆地原油样品中芳烃化合物亚组分“两步法”检测结果Table 2 Results of “two-step” separation for aromatic sub-fractions in crude oil samples from Termit Basin
如图3所示,比较石油醚/二氯甲烷(9/1)试剂分离出的双环多环芳烃系列和石油醚/二氯甲烷(8/2)的试剂分离出的三环多环芳烃系列的总离子流图可以看出,在保留时间21~22 min之间,一些C2-二苯并呋喃化合物似乎在两种不同极性冲洗下来的馏分中都存在,有理由认为该种化合物同时能克服以上两种配比试剂的极性区间。由于氧芴类化合物是由两个苯环和一个五元环组成,此类化合物的极性比双环系列里的萘要强一些,比三环系列里的菲要弱一些,所以C2-二苯并呋喃同时出现在了双环组分的结尾和三环组分的开头。
如表2所示,石油醚/二氯甲烷(95/5)和(9/1)都洗脱出了萘系物,与“一步法”结果一样,前者洗脱的浓度很低,因此使用石油醚/二氯甲烷(9/1)溶剂洗脱双环芳烃系列化合物并进行单体碳同位素的测定更加可靠。如图4所示,使用石油醚/二氯甲烷(8/2)溶剂能将菲系物,芴系列,氧芴系列和硫芴系列[32-33]分离开,而且多种单体化合物是单独出峰,很大程度上减少了其他化合物的干扰,这样的效果对于芳香烃中个别单体化合物碳同位素分析有着重要的意义。与“一步法”相比,“二步法”较好地分开了萘系物和菲系物,并且这两类化合物在两种亚组分中不存在分馏现象,实验结果表明3个油样有着相同的规律,这样的效果对碳同位素的测定影响不大,因此,可以认为萘系物与菲系物分离得非常清楚,此种方法在分离双环多环芳烃系列和三环多环芳烃系列时非常可靠。
3.3 “两步法”分离技术的适用性
“二步法”分离技术是在得到芳烃族组分后,再进行第二次精细分离得到不同环数的芳烃化合物亚组分。而无论是海相原油、陆相原油、高成熟凝析油还是正常原油都含有一定量的饱和烃、芳烃、非烃和沥青质,只是比例上存在很大的差异。在原油定量的情况下,芳烃的质量有一个取值范围(0~60 mg),而6 mL的试剂足以将目标化合物冲洗完毕[21],因此,目标化合物不会因为占比多而不能完全被冲洗下来。如果占比较少,甚至为0的话,也不会因为目标化合物含量低而提前把其他的化合物冲洗出来,因为针对每一种的目标系列化合物都采用不同的极性配比,且冲洗剂极性是由弱变强的,极性弱的试剂即使用量过度也不会将非目标物质提前冲洗下来。因此,此方法可以有效克服原油性质的影响,如族组分的相对比例,成熟度等因素的影响。事实上,如果轻质原油中的极性物质含量很少,那么可能一步法也可以实现(因为上层的氧化铝没有失效),但二步法提出了一种相对标准的流程,可以避免因样品差异性导致的问题,有利于该方法的推广。在某种程度上,冲洗剂的体积与原油的质量成正相关。本文选取的尼日尔Termit盆地的3个样品都检测出相似的结果,因此,可以证明此方法的适用性。
使用“两步法”对芳烃馏分进行精细分离亚馏分后,如图4所示,发现石油醚/二氯甲烷(8/2)分离出的馏分的UCM比芳烃总馏分的高度变低了。一般认为在化合物的浓度相差不大时基线会保持平稳状态,当保留时间靠后的化合物的浓度比靠前的浓度低出很多时,会造成基线的漂移。图4b是使用“两步法”把单环芳烃、双环芳烃分离出去后,对石油醚/二氯甲烷(8/2)分离出的馏分做GC-MS,相当于对前面单环芳烃和双环芳烃部分的基线进行了清空,因此,UCM的高度会变低,出现的时间也会相对延后。
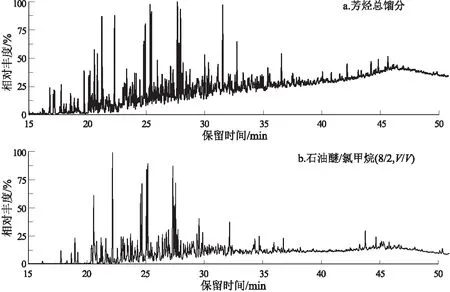
图4 盆地原油样品芳烃总馏分与“两步法”中使用石油醚/二氯甲烷试剂洗脱出的化合物总离子流图Fig.4 Total aromatic fractions and total ion flow diagrams of compounds eluted with petroleum ether/dichloromethane solvent in “two-step” process of crude oil samples from Termit Basin
4 结论
(1)在“两步法”中使用石油醚/二氯甲烷(99/1)的试剂冲洗出的第一亚馏分,经GC-MS检测发现总离子流图呈等间距分布,识别判断为烷基苯系列化合物,但只能根据质量数判断出化合物的分子式。
(2)使用石油醚/二氯甲烷(9/1)比(95/5)配比的试剂冲洗下来的双环芳烃系物浓度大出10倍,由于GC-IR-MS分析不仅需要基线分离,对浓度也有一定的标准,所以就不同极性的洗脱剂而言,石油醚/二氯甲烷(9/1)更适合冲洗双环多环烃系物的化合物。
(3)在“两步法”中使用第二步分离时,石油醚/二氯甲烷(9/1)和(8/2)两种配比试剂可以将双环芳烃化合物和三环芳烃化合物有效分开,有效解决萘系物和菲系物同分异构体共流出问题,达到GC-IR-MS检测标准,获取大部分二环和三环芳烃单体烃碳同位素组成特征。
猜你喜欢
石油沥青(2022年4期)2022-09-03
石油炼制与化工(2022年6期)2022-06-21
安徽农学通报(2022年8期)2022-05-06
炼油技术与工程(2022年4期)2022-04-20
化学工程师(2022年3期)2022-04-19
石油学报(石油加工)(2022年2期)2022-03-11
上海化工(2021年2期)2021-04-23
家用汽车(2016年12期)2017-02-09
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
