
首发于中枢神经系统的急性显微镜下多血管炎一例及报告
2021-06-02 01:20吴玉萍张洪
中华卫生应急电子杂志 2021年2期
吴玉萍 张洪
作者单位:430071 湖北武汉,武汉大学中南医院神经内科
显微镜下多血管炎(microscopic polyangiitis,MPA)与肉芽肿性多血管炎和嗜酸性肉芽肿性多血管炎一起构成抗中性粒细胞胞浆抗体(antineutrophil cytoplasmic antibodies,ANCA)相关性血管炎[1]。研究发现[2],MPA多见于欧美国家,临床表现复杂多样、无特异性,以肺部及肾脏受累最为常见。虽然近年来国内也有报道累及肺部和肾脏的MPA病例,但以中枢神经系统为首发症状的MPA病例罕见。笔者报告一例武汉大学中南医院神经内科收治的累及中枢神经系统、以急性脑梗死为首发症状的MPA患者。
一、病例资料
患者男性,29岁,因“右侧肢体麻木无力7 d、加重2 d”于2019年8月3日入院。患者在入院1周前突然出现右侧手脚乏力、活动困难。右上肢表现为不能写字、不能使用鼠标;右下肢表现为跛行;伴有右侧肢体麻木不适。入院2 d前患者症状加重,右手握不住筷子,右上肢不能平举,右下肢无法行走。发病过程中无发热、咳嗽、头痛、呕吐、视物模糊和意识障碍,大小便正常。既往史中无高血压、高血脂、糖尿病和心脏病病史,近期无疫苗接种史,没有抽烟、饮酒、喝茶及喝咖啡等嗜好。入院体检:体温36.8℃,呼吸20次/min,脉博69次/min,血压131/81 mmHg(1 mmHg=0.133 kPa)。心肺腹部体检未见异常。神经系统检查:神清,右侧鼻唇沟较左侧浅,颈软,右上下肢肌力Ⅲ级,左上下肢肌力Ⅴ级,右上下肢痛温觉较左侧减退,双侧肢体病理征阴性。脑部CT可见左侧基底节区低密度病灶(图1),头颈部CTA无异常,以“急性脑梗死”收住院诊治。入院后辅助检查结果提示:三大常规和血生化全套未见异常;风湿、类风湿全套、凝血象全套、传染病四项、肿瘤标志物正常。免疫学检查发现血蛋白酶3抗体阳性。淋巴细胞亚群结果提示T淋巴细胞增高(72.26%),辅助/诱导性T淋巴细胞增高(40.47%),B淋巴细胞正常(22.88%),NK细胞下降(3.50%)。心脏超声波检查没有异常发现。入院后3 d脑部MRI+DWI+ADC提示左侧基底节异常信号(图2);入院后5 d脑灌注增强和波谱成像提示左侧基底节区病灶边缘少许中度强化(图3a),考虑为低级别胶质瘤或感染性病变可能。胸部、腹部及盆腔CT未见异常;全脊柱MRI平扫和增强未见异常。入院后7 d脑脊液结果外观正常,压力正常;蛋白质含量稍高(0.54 g/L)、糖和氯化物含量正常,细胞数正常;细胞染色正常;脑脊液电泳未见寡克隆带。入院后12 d在神经外科医师协助下,用脑立体定向技术对左侧基底节区进行活检,穿刺组织为脑白质成分,可见反应性胶质细胞增生及散在组织细胞,小血管见广泛钙化,部分血管壁破坏及炎性细胞构成的套状结构;结合免疫组化和特殊染色结果考虑病理诊断为血管炎伴血管壁钙化(图4)。全身PET-CT未见到明显恶性肿瘤病变征象,左侧基底节区低密度灶、代谢不高,考虑多为ANCA相关性血管炎所致改变。本例患者临床诊断为MPA。治疗上停用脑梗塞药物;予以甲强龙冲击治疗,初始剂量为500 mg、1次/d静脉滴注,每3 d剂量减半,当剂量减至60 mg时改为强的松口服治疗;静脉滴注免疫球蛋白20 g/d、连续5 d;环磷酰胺600 mg 静脉滴注1次。入院后40 d复查脑部MRI发现左侧基底节区病灶边缘强化较前缩小(图3 b)。患者右侧肢体肌力达到IV级,于入院后53 d出院,随诊患者病情稳定。
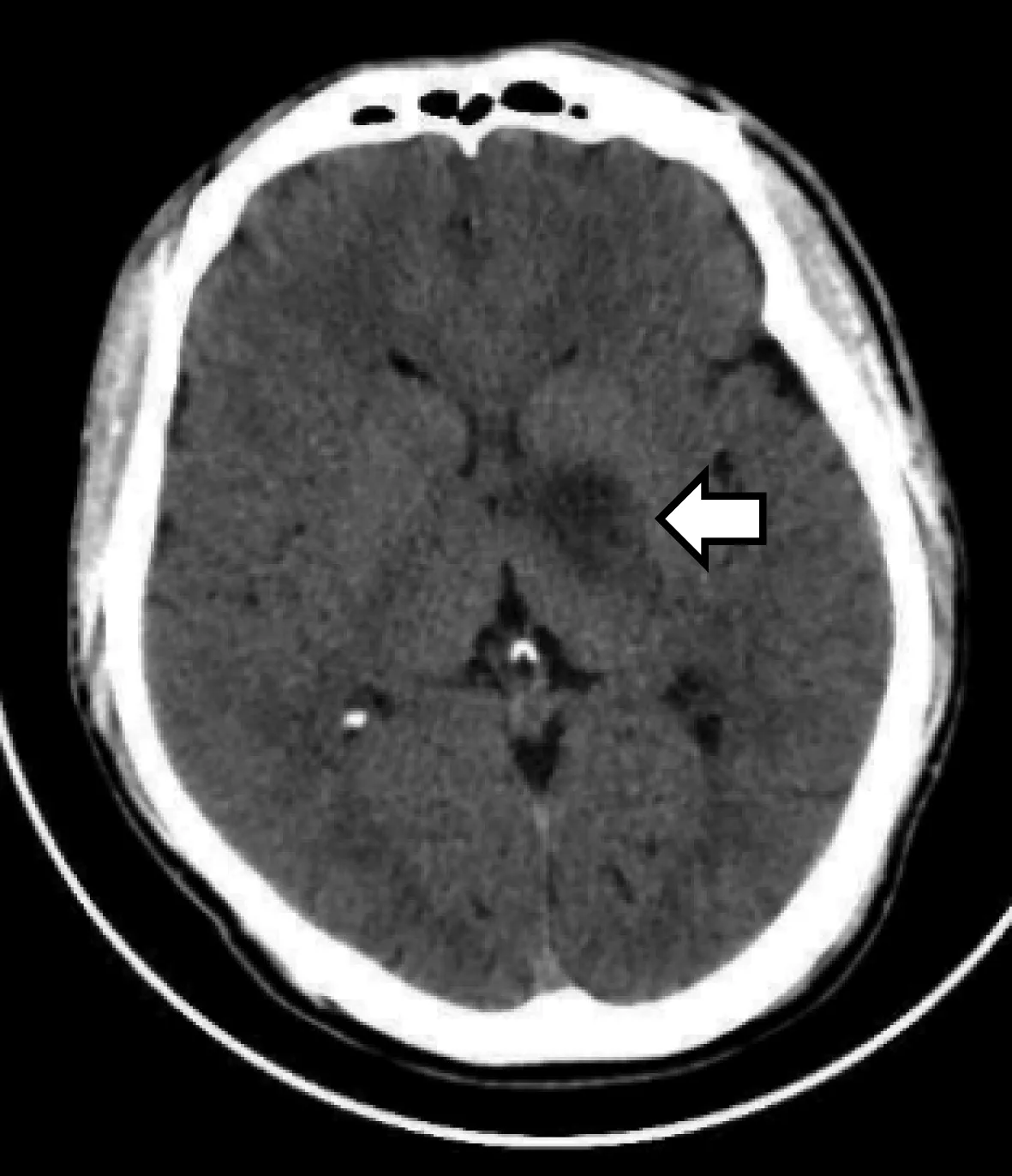
图1 头部CT提示左侧基底节区低密度灶
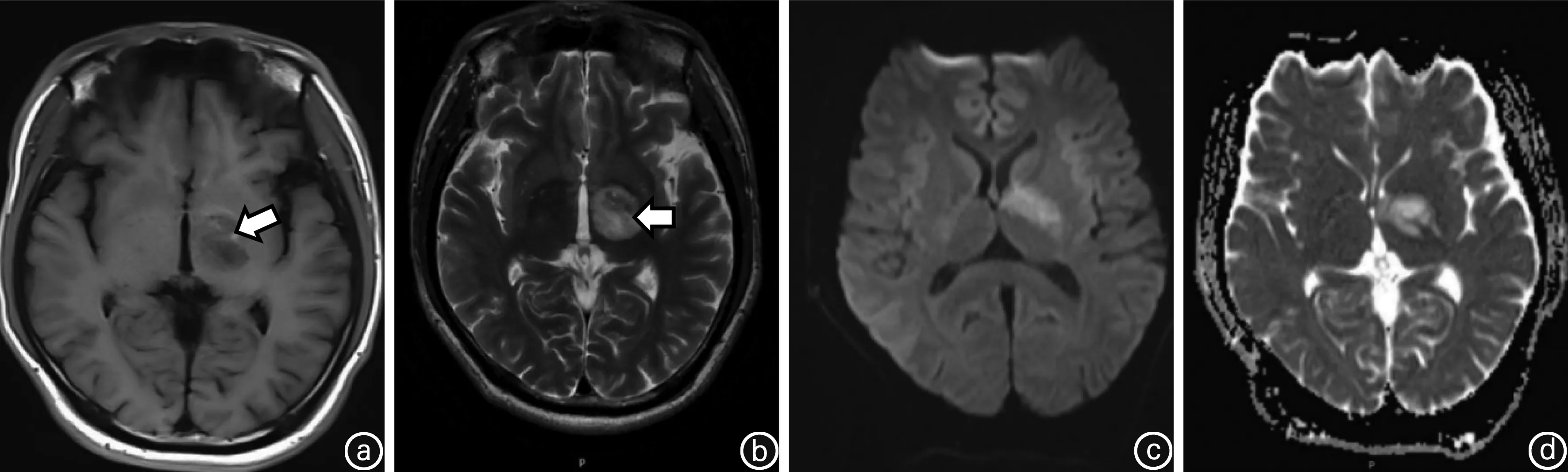
图2 入院后3 d头部MRI影像图

图3 头部MRI增强影像比较

图4 左侧基底节区脑组织活检病理结果(HE染色 10×40)
二、讨论
1923年Wohlwill[3]首次将两名患者描述为“结节性周围性动脉炎的显微镜下改变”,这种疾病现在称为MPA,是一种原发性系统性血管炎,其特征是小口径血管的炎症和循环中中性粒细胞胞浆抗体的存在。1985年Savage等[4]首次提出MPA是一种以系统性血管炎为特征的特发性自身免疫疾病。MPA的发病率低,以男性为主,其病因未明,有家族遗传倾向[5]。虽然MPA是由临床和病理学的标准来定义,研究发现近80%的MPA患者可检测到抗蛋白酶3(proteinase 3,PR3)抗体、或抗髓过氧化物酶(myeloperoxidase,MPO)抗体。病理学研究发现[2],在MPA中最常受影响的器官是肾脏和肺,迅速进行性肾衰竭可显示坏死性和新月性肾小球肾炎。对已发表的累及肾脏的文献进行分析发现[2],MPA临床上容易被误诊为肾盂肾炎、肾综合征出血热、慢性肾小球肾炎、急性间质性肾炎、过敏性紫癜性肾炎、慢性肾功能不全或尿毒症。通过分析已发表的文章发现[2],累及肺部的MPA容易误诊为上呼吸道感染、支气管肺炎、慢性支气管炎、支气管扩张症、肺部感染、间质性肺炎、重症肺炎、肺炎并心力衰竭、社区获得性肺炎、肺部炎症合并肺间质纤维化、肺部炎症合并胸膜增厚、慢性阻塞性肺疾病急性加重、肺结核、咳嗽变异性哮喘、肺出血-肾炎综合征。值得一提的是,在国外研究中[6-7],虽然皮肤、胃肠道、耳、鼻和喉的表现常不被认为是MPA的主要症状,但在多数患者的描述中发现耳、鼻和喉部症状的比例出乎意料的高。国外的研究报道以中枢神经系统受累为主要表现的是脑血管意外[8],如蛛网膜下腔出血[9-10]、脊髓硬膜内出血和脑出血[11-12]、脑梗死[13]、桥脑梗死[14]、延髓梗死[15]、颈脊髓炎[16]。
在国内文献中,有些MPA患者被误诊为急性阑尾炎、消化道出血、慢性胃炎、 急性胃肠炎、肝炎、关节炎和贫血[17-18]。国内MPA患者表现为中枢神经系统受累的病例报道很少。2008年张文等[19]首次在国内期刊描述3例MPA患者出现中枢神经系统损害,表现为脑实质病变,没有病理检查结果。2014年包国庆等[20]报告1例MPA患者以言语不清、左侧肢体偏瘫,左侧偏身麻木为表现,曾诊断为多发性脑缺血,后经肾脏穿刺病理学检查诊断MPA。2018年段红莉等[21]报告1例MPA患者,表现为脑梗死、合并左侧桡神经损害,患者未做病理活检。本例患者以急性右侧肢体偏瘫为首发症状,头部CT提示左基底节区低密度灶,接诊时疑似“急性脑梗死”的诊断。
目前MPA尚无统一的诊断标准,临床诊断主要由临床表现、影像学检查、ANCA抗体检测以及肾和肺活检来确定[22-23]。诊断要点:(1)中老年男性多见。(2)累计肾脏、肺、消化道、皮肤、关节、神经等多个系统的症状或体征。(3)实验室或影像学检查提示各器官系统损害的证据。(4)ANCA检测阳性。(5)肾脏或肺活检提示小血管炎的病理改变。针对中枢神经系统的病变,脑组织活检对脑部病灶明确诊断具有关键性的作用,抗PR3/C-ANCA 抗体阳性可资MPA的临床诊断。有研究发现[24],在MPA的患者中,IgM PR3-ANCA抗体的检测率为41.1%,且与疾病严重程度相关。另有文献报道[25],在MPO-ANCA血管炎中常看到皮肤和肾脏损害的表现,而在PR3-ANCA血管炎中常有黏膜、眼、耳鼻喉和肾脏损害表现,但在3个AAV类别之间,肺和神经系统表现无明显差异。本例患者为年轻男性,既往无特殊病史,患者没有出现肺部和肾脏异常表现,在排除心源性、血液源性、动脉粥样硬化性、风湿类风湿性以及脱髓鞘性病变和肿瘤外,通过核磁共振影像或PET扫描结果,ANCA抗体的检测以及脑部组织病理活检,最终诊断为MPA。MPA标准化的治疗方案基于环磷酰胺和皮质类固醇,使用这种方案,可以使大多数患者缓解。本例患者在明确诊断后,得到标准化的治疗,出院时患者症状明显好转,右侧肢体肌力得到改善,头部核磁共振的病灶也有缩小。目前患者病情稳定。
通过对本例MPA患者的诊疗过程,笔者认为临床一线医师应注意如下问题:(1)要充分认识MPA患者复杂的临床表现,该病常表现为身体多器官系统受累的非特异性症状,亦有少数仅累及一个系统。(2)MPA可在任何年龄段发病,可急性或者隐匿起病。(3)对于病情复杂、诊断困难的患者,或用常规治疗方案而临床疗效较差的患者,要考虑自身免疫性疾病的可能,积极完善相关的免疫学检查。(4)及早进行病理活检有助于MPA的确诊。尽量避免MPA的临床误诊误治,做到早期诊断和早期治疗,减少疾病残疾的发生,让患者重返工作或学习生活,减轻家庭及社会负担。
总之,MPA临床表现极为复杂,影像学表现无特异性,容易误诊。临床医师需提高对MPA的警惕性,拓宽诊断思路,才能减少误诊,缩短确诊时间。及早应用糖皮质激素和免疫抑制剂治疗,才能控制病情,实现良好预后。
猜你喜欢
传染病信息(2022年2期)2022-07-15
中国典型病例大全(2022年9期)2022-04-19
现代临床医学(2022年1期)2022-02-12
中国典型病例大全(2022年1期)2022-01-10
昆明医科大学学报(2021年1期)2021-02-07
健康体检与管理(2021年10期)2021-01-03
中外医疗(2018年12期)2018-09-03
中西医结合心血管病电子杂志(2016年23期)2017-03-03
中国实用医药(2016年30期)2016-12-28
小星星·阅读100分(高年级)(2014年9期)2014-09-23
