
6种氯代乙烯阳离子的密度泛函理论研究
2021-05-27 09:18:40张晓琳焉炳飞刘绍丽李文佐
烟台大学学报(自然科学与工程版) 2021年2期
张晓琳,焉炳飞,刘绍丽,李文佐
(烟台大学化学化工学院,山东 烟台 264005)

氯代乙烯分子失去一个电子后得到阳离子。实验上,人们采用多种光谱技术如红外光谱、拉曼光谱、光电子能谱、共振加强多光子电离光谱、激光诱导荧光光谱、电子自旋共振等对它们进行研究, 能够得到大量信息。然而,实验无法直接给出阳离子的电子基态,一般也不能给出离子的结构参数,对光谱结果的解释和归属及各种反应机理的阐释离不开量子化学计算[3]。本课题组对6种氯代乙烯阳离子进行了理论计算,指认了它们的电子基态,计算了对应分子的垂直电离势(VIP)和绝热电离势(AIP),并与实验结果进行了对比,为全面认识和理解氯代乙烯阳离子的结构和性质提供了有用信息。
1 计算方法
计算主要采用密度泛函理论B3LYP[4-5]方法,选择6-311G(d,p)、6-311++G(d,p)和cc-pVTZ基组[6](3种基组分别表示为B1,B1+和B2),对所有6种氯代乙烯阳离子和对应的分子进行构型优化并计算振动频率以确定优化得到的构型是否对应势能面上的最低点。为了比较,同时采用QCISD[7-8]方法在相同基组上进行计算。对各个阳离子在相同计算水平上进行了自然布居分析计算[9]。计算的VIP值为在分子的优化构型上计算的离子(基态)和分子的能量差,AIP值为离子(基态)和分子在各自的优化构型上的能量差。所有计算均使用Gaussian09程序[10]。
2 结果与讨论
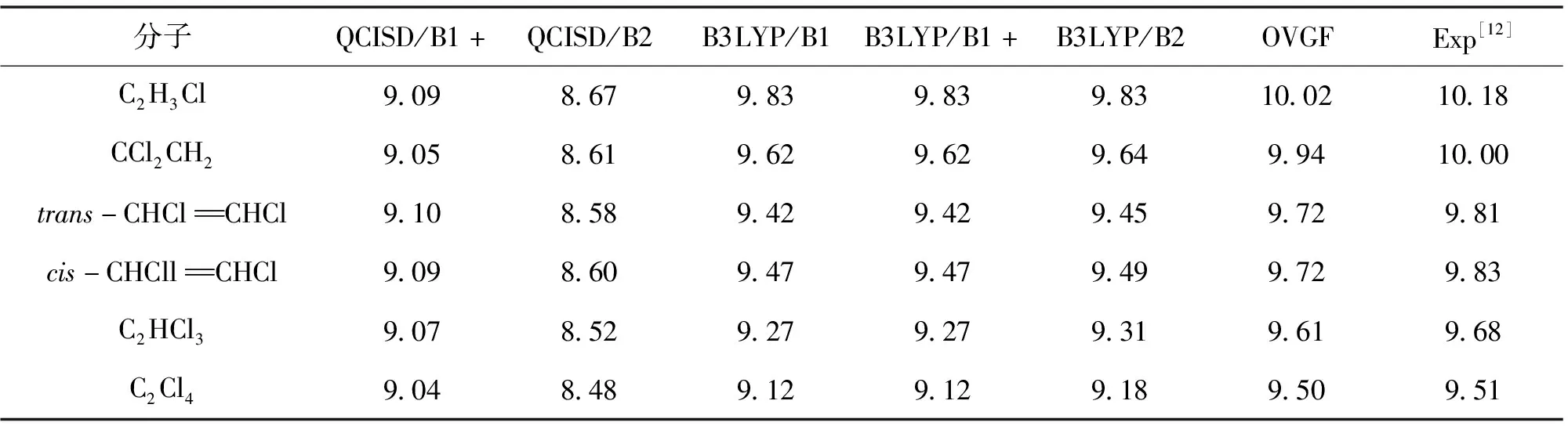
表2 用B3LYP和QCISD方法计算的氯代乙烯分子的垂直电离势
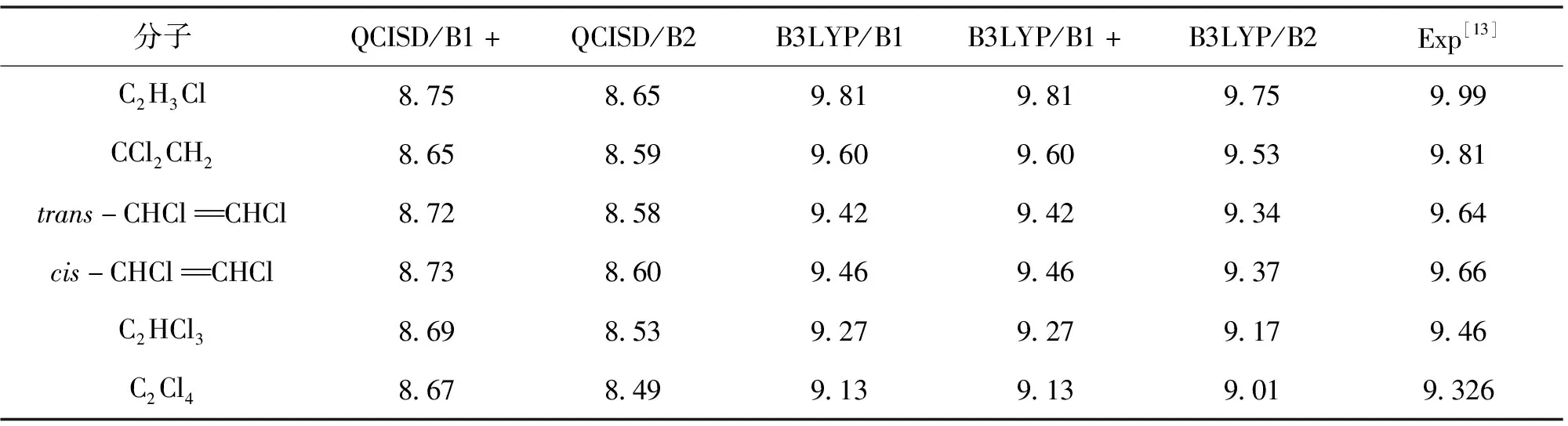
表3 用B3LYP和QCISD方法计算的氯代乙烯分子的绝热电离势
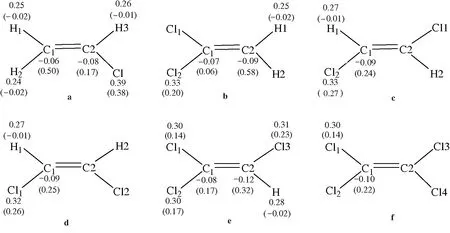
图1 6种氯代乙烯阳离子的自然电荷布居(括号内为自旋密度值)
2.1 氯代乙烯阳离子的结构
计算结果表明,同相应的分子一样,6种氯代乙烯阳离子都具有平面结构。各个级别的频率计算表明,6种离子的优化构型都对应于势能面上的最低点,即为稳定结构。研究[11]表明,乙烯阳离子具有非平面结构(D2对称性)。我们曾参照乙烯阳离子的结构进行构型优化计算,但最后都只能得到平面结构。这表明氯代的乙烯阳离子都只具有平面结构。计算结果显示氯代乙烯阳离子a、b、c、d、e和f分别具有Cs、C2v、C2h、C2v、Cs和D2h对称性,它们的电子基态分别为2A"、2B1、2Au、2B1、2A"、2B3u,全部是“π型”自由基。
关于离子的优化构型,本研究主要讨论B3LYP/B1, B3LYP/B1+,B3LYP/B2级别上的计算结果(表1)。
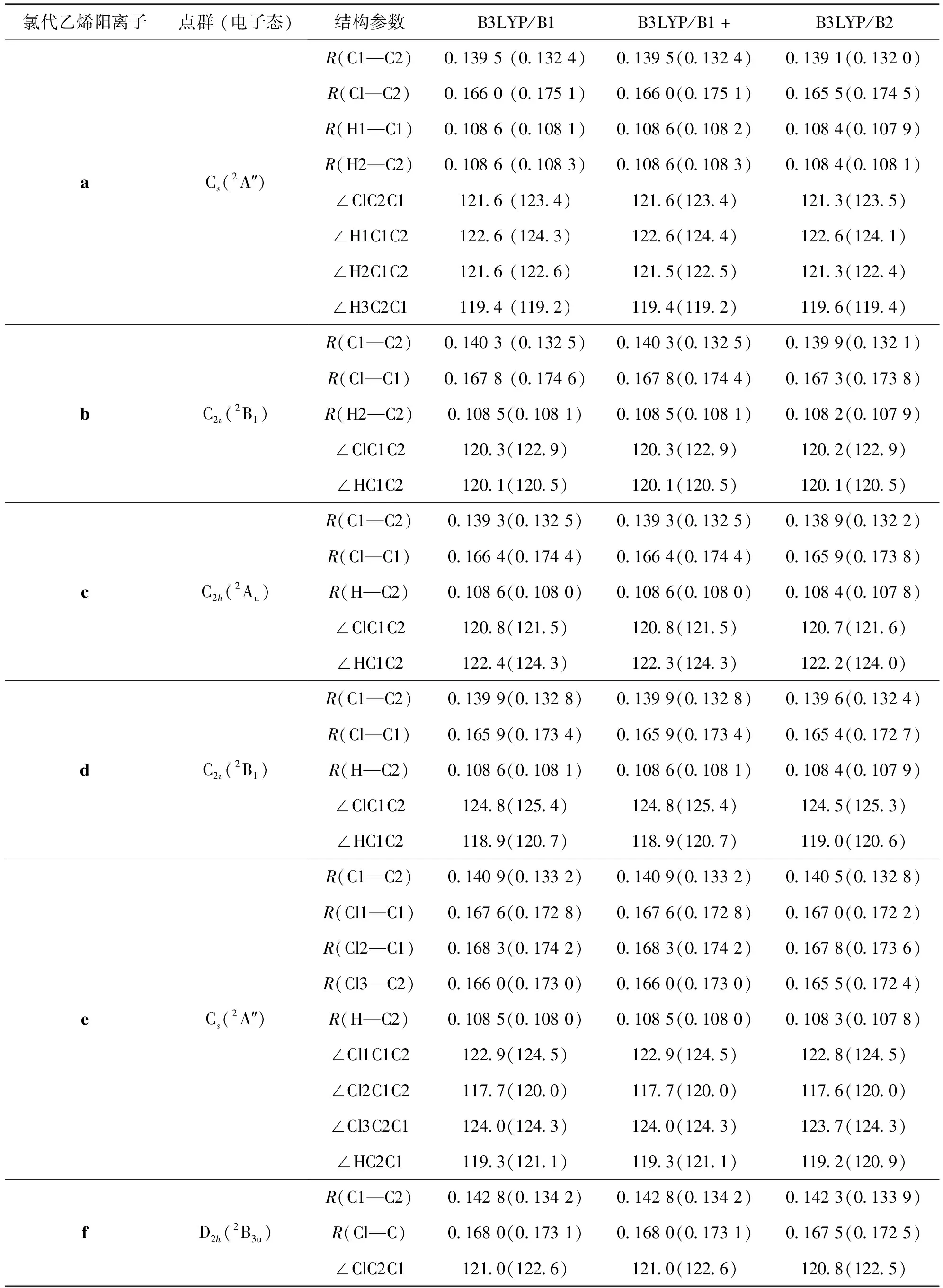
表1 用B3LYP方法及B1、B1+和B2基组计算的6种氯代乙烯阳离子(a—f)的结构
比较计算得到的氯代乙烯阳离子和对应分子的结构发现,在B3LYP/B1和B3LYP/B1+级别上计算的构型参数值非常接近,在这2个级别上的键长计算值相差不超过0.000 5 nm,键角值相差不超过0.2°。这说明在基组中包含弥散基函数(“+”)对所计算的离子和分子的构型参数值的影响不大。
2.2 氯代乙烯阳离子的自然布居分析
各氯代乙烯阳离子的自然电荷数据(图1)表明,这些阳离子的正电荷都分布在Cl原子和各H原子上,所有的C原子上分布着负电荷。
各氯代乙烯阳离子中的自旋密度数据(图1)表明,自旋密度主要分布在Cl原子和C原子上,H原子上自旋密度值很小(H上有很小的负值)。
2.3 氯代乙烯分子的垂直电离势(VIP)和绝热电离势(AIP)
检验计算阳离子时各个计算级别上的S2算子的本征值发现,用B3LYP方法计算时,
从表2可以看出,B3LYP计算的VIP值与相应实验值误差大约在0.4 eV,可以认为应用B3LYP方法能够较合理地预言氯代乙烯分子的垂直电离势。我们还用更高级的外价层格林函数方法(OVGF)计算了所有氯代乙烯阳离子的VIP(表3),OVGF计算的VIP与相应实验值非常接近(误差小于0.16 eV),说明该方法能够精确预言氯代乙烯阳离子的垂直电离势。从表3可以看出,B3LYP计算的AIP值与相应实验值误差大约在0.2 eV,可以认为应用B3LYP方法能够较准确地预言氯代乙烯分子的绝热电离势。由表2和表3可知,QCISD计算的VIP和AIP结果误差很大;此外,用B1和B1+基组计算的VIP和AIP值基本一致,说明弥散基的加入并不能改善对该类分子的垂直电离势和绝热电离势的计算,这与以往对氟代乙烯分子的电离势的计算结果[14]不同。
3 结 论
猜你喜欢
中学生数理化·自主招生(2022年10期)2022-05-30 10:48:04
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27 03:06:58
今日农业(2019年11期)2019-08-13 00:49:02
火工品(2019年6期)2019-06-05 02:35:44
厦门大学学报(自然科学版)(2018年2期)2018-04-11 07:07:35
价值工程(2017年31期)2018-01-17 00:49:24
中国司法鉴定(2017年5期)2017-10-11 02:36:45
中学教学参考·理科版(2016年3期)2017-05-19 18:38:46
当代化工研究(2016年5期)2016-03-20 16:21:32
环境科技(2015年2期)2015-11-08 12:11:22
