
Gut microbiota dysbiosis in Chinese children with type 1 diabetes mellitus: An observational study
2021-05-25 10:07XiaLiuYiWenChengLiShaoShuHongSunJianWuQingHaiSongHongShengZouZongXinLing
World Journal of Gastroenterology 2021年19期
Xia Liu, Yi-Wen Cheng, Li Shao, Shu-Hong Sun, Jian Wu, Qing-Hai Song, Hong Sheng Zou, Zong-Xin Ling
Abstract
Key Words: Dysbiosis; Fasting blood glucose; Sequencing; Metabolism; Type 1 diabetes mellitus
INTRODUCTION
Type 1 diabetes mellitus (T1 DM), a chronic autoimmune disease that usually begins in childhood, results from the destruction (or loss) of pancreatic β-cells, which leads to an inability to produce insulin and a need for the administration of exogenous insulin.T1 DM ranks as the second most common autoimmune disease among children. The peak incidence of T1 DM is clearly within the age group of 10 –14 years and declines thereafter. One nationwide, population based study demonstrated that the incidence of T1 DM in children is 1 .93 (ranging from 0 .83 to 3 .03 ) per 100000 person-years in China, with an annual increase of approximately 6 .5 %[1 ]. T1 DM can cause lifechanging and life-threatening health complications in many organs and tissues rich in capillary vessels, such as the kidney, retina, and nerves, resulting in premature death.Children with T1 DM often have to be treated for hypertension, dyslipidemia, microalbuminuria, and nephropathy, among other conditions[2]. Its early onset and chronicity make T1 DM a disease of considerable importance. Currently, T1 DM and its related comorbidities are considered major public health concerns.
Over the last few decades, considerable progress has been made in research on the pathogenesis and treatment of T1 DM. However, its precise etiology and pathological mechanisms remain largely unclear. Both genetic susceptibility and environmental factors contribute to the development of T1 DM[3 ]. The genetic susceptibility associated with T1 DM is fairly well known, whereas the environmental factors remain poorly defined despite intensive research. Among the environmental risk factors associated with T1 DM are industrial and economic advances (such as high levels of hygiene), changes in diet, and the emergence of more sedentary lifestyles[4], which can affect the environmental exposure of children. This altered environmental exposures can directly and indirectly influence the early-life gut microbiota.
Numerous clinical and experimental reports provide growing evidence of a close link between an altered gut microbiota (also defined as dysbiosis) and T1 DM[5 -12 ]. A previous study found that T1 DM-related dysbiosis is associated with reduced integrity and increased permeability of the gut mucosa, which leads to bacterial penetration,and can stimulate the immune system to produce antibodies. Cross-reaction of these antibodies and surface antigens of pancreatic beta cells, as well as T cell crossreactivity, results in the destruction of pancreatic β-cells and the development of T1 DM[13 ]. Specifically, the Firmicutes/Bacteroidetes ratio is significantly reduced in Finnish children with T1 DM[14 ]. Leiva-Gea et al[6 ] demonstrated thatBacteroidesandVeillonellaare markedly enriched in patients with TIDM, whereasFaecalibacteriumandRoseburiaare significantly reduced[6 ]. Livanos et al[11 ] showed that early-life antibiotic treatments alter the gut microbiota and its metabolic capacities, intestinal gene expression, and T-cell populations, thereby accelerating T1 DM onset in non-obese diabetic mice[11 ]. Taken together, these findings show that early dysbiosis of the gut microbiota can be used as a promising biomarker for T1 DM.
Most of the previous studies on the T1 DM-associated microbiota were conducted in the United States and Europe. However, differences in lifestyle, dietary constitution,environmental exposures, and host genetic background between Chinese and Western populations may contribute to disparities in the baseline microbiota composition,which may influence the roles of specific bacteria in the etiopathology of T1 DM. The present study aimed to explore the T1 DM-associated gut microbiota in Chinese children by using the 16 S rRNA gene high-throughput sequencing platform. We also evaluated the correlation between the T1 DM-associated gut microbiota and fasting blood glucose (FBG). Our findings suggest novel targets for noninvasive early diagnosis and personalized treatment of T1 DM.
MATERIALS AND METHODS
Participant selection
A total of 51 children with newly confirmed T1 DM (aged 6 -14 years) and 47 age-,gender-, and education-matched healthy controls were enrolled from Linyi People’s Hospital (Linyi, China) and the First Affiliated Hospital, School of Medicine, Zhejiang University (Hangzhou, China) from June 2019 to November 2019 . The diagnosis of T1 DM was based on the criteria of the American Diabetes Association: T1 DMassociated autoimmunity (i.e., formation of islet autoantibodies); the classic trio of symptoms associated with disease onset,i.e.,polydipsia, polyphagia, and polyuria along with hyperglycemia; an immediate need for exogenous insulin replacement and lifetime treatment. All the T1 DM children were treated with only insulin. The FBG level of these participants was determined in the morning (Table 1). The following exclusion criteria were established: Body mass index (BMI) ≥ 30 kg/m2 ; use of antibiotics, probiotics, prebiotics, or synbiotics in the previous month; known active infections such as bacterial, fungal, chlamydial, or viral infections; and other diseases such as irritable bowel syndrome (IBS), inflammatory bowel disease or other autoimmune diseases. The protocols for the present study were approved by the Ethics Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University. Informed written consent was obtained from the subjects’ guardians beforeenrollment.

Table 1 Fundamental information of subjects
Fecal sample collection and microbial DNA extraction
According to our previous studies, approximately 2 g of fresh fecal sample was collected in a sterile plastic cup, and stored at -80 °C within 15 min after preparation until use. Bacterial genomic DNA was extracted from 300 mg of homogenized feces using a QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, with additional glass-bead beating steps on a Mini-Beadbeater (FastPrep; Thermo Electron Corporation, Boston, MA, United States). The amount of DNA was determined using a NanoDrop ND-1000 spectrophotometer(Thermo Electron Corporation); the integrity and size were checked by 1 .0 % agarose gel electrophoresis on a gel containing 0 .5 mg/mL ethidium bromide. All DNA was stored at -20 °C before further analysis.
Amplicon library construction and sequencing
Amplicon libraries were constructed with the Illumina sequencing-compatible and barcode-indexed bacterial polymerase chain reaction primers 338 F (5 ’-ACTCCTRCGGGAGGCAGCAG-3 ’) and 806 R (5 ’-GGACTACCVGGGTATCTAAT-3 ’), which targeted the V3 -V4 regions of 16 S rRNA gene. All PCRs were performed with KAPA HiFi HotStart ReadyMix using the manufacturer's protocol (KAPA Biosystems,Wilmington, MA) and approximately 50 ng of extracted DNA was used per reaction.Thermocycling conditions were as follows: 30 cycles of 95 °C for 1 min, 55 °C for 1 min,and 72 °C for 1 min, followed by a final extension at 72 °C for 5 min. All PCRs were performed in triplicate in a volume of 50 mL, and the samples were combined after PCR. The amplicon library was prepared using a TruSeqTMDNA Sample Preparation Kit (Illumina Inc, San Diego, CA, United States). Prior to sequencing, the PCR products were extracted with the MiniElute Gel Extraction Kit (Qiagen) and quantified on a NanoDrop ND-1000 spectrophotometer (Thermo Electron Corporation) and Qubit 2 .0 fluorometer (Invitrogen). The purified amplicons were then pooled in equimolar concentrations and the final concentration of the library was determined by Qubit(Invitrogen). Negative DNA extraction controls (lysis buffer and kit reagents only)were amplified and sequenced as contamination controls. Sequencing was performed on a MiSeq instrument (Illumina) using a 300 × 2 V3 kit together with PhiX Control V3(Illumina)[15 ,16 ].
Bioinformatic analysis
The 16 S rRNA gene sequence data set generated from the Illumina MiSeq platform was inputted to QIIME2 (version 2020 .11 ), and all steps of sequence processing and quality control were performed in QIIME2 with default parameters[15 ,16 ]. Before the following data analysis, these reads of each sample were normalized to even sampling depths and annotated using the Greengenes reference database (version 13 .8 ) with both the Ribosomal Database Project Classifier and UCLUST version 1 .2 .22 methods implemented in QIIME. Alpha diversity, including the observed species, abundancebased coverage estimator (ACE), Chao1 estimator, Shannon, Simpson, Evenness, and PD whole tree indices, was calculated at a 97 % similarity level. Beta diversity was measured by the unweighted UniFrac, weighted UniFrac, Jaccard, and Bray-Curtis distances calculated by QIIME2 , which were visualized by principal coordinate analysis (PCoA). The differences in the composition of the fecal microbiota at different taxonomic levels were analyzed with Statistical Analysis of Metagenomic Profiles(STAMP) software package v2 .1 .3 and by the linear discriminant analysis effect size(LEfSe) method. PiCRUSt v1 .0 .0 was used to identify predicted gene families and associated pathways from inferred metagenomes of taxa of interest identified from the compositional analyses. The sparse compositional correlation (SparCC) algorithm was used for correlation analysis, and the results were visualized using Cytoscape v3 .4 .1 .
Statistical analysis
For continuous variables, independentt-tests, White’s nonparametrict-tests, and Mann-WhitneyU-tests were applied. For categorical variables between groups,Pearson’s chi-square test or Fisher’s exact test was used, depending on assumption validity. For correlation analyses, Spearman’s rank correlation test was used. Statistical analyses were performed using SPSS V19 .0 (SPSS Inc., Chicago, IL, United States) and STAMP V2 .1 .3 . GraphPad Prism version 6 .0 (San Diego, CA, United States) was used to prepare graphs. All tests of significance were two sided, andP< 0 .05 or correctedP< 0 .05 was considered statistically significant.
Accession number
The sequence data from this study are deposited in the GenBank Sequence Read Archive with the accession number SRP287193 .
RESULTS
Altered bacterial diversity in children with T1 DM
No significant differences were noted in age, gender, race, BMI, or lipid levels between Chinese children with T1 DM and healthy controls (P > 0 .05 ). The FBG level was significantly higher in children with T1 DM than in healthy controls (Figure 1 ;P<0 .05 ). A total of 3961145 high-quality reads (1857135 reads in healthy controls and 2104010 reads in children with T1 DM) with an average of 40420 reads per sample were obtained for subsequent microbiota analysis. Deeper sequencing identified the majority of the bacterial phylotypes [640 operational taxonomic units (OTUs)] present in the fecal microbiota. The Good’s estimator of coverage was 99 .93 %.
For bacterial diversity analyses, the Shannon and Simpson indices differed significantly between children with T1 DM and healthy controls (P < 0 .05 ; Figure 2 A and B), with an increased diversity in the T1 DM-associated fecal microbiota. However,richness indices, such as the ACE and Chao1 indices, showed no significant changes between the two groups (P> 0 .05 ; Figure 2 C and D). Owing to the significant interindividual variations, PCoA based on unweighted UniFrac, weighted UniFrac, and Bray–Curtis algorithms could not separate the two groups into different clusters(Figure 2 E-G). Based on the Venn diagram in Figure 2 H and Shannon rarefaction curves (Figure 2 I), we observed a slightly higher number of OTUs in children with T1 DM. Taken together, the results of our deeper sequencing analysis indicated increased fecal microbial diversity in children with T1 DM.
Altered fecal microbiota composition in children with T1 DM
The overall microbial compositions in children with T1 DM and healthy controls were examined at different taxonomic levels from phylum to species. Using the RDP Classifier, sequences were annotated as follows: 11 phyla, 29 orders, 53 families, 184 genera, and 271 species. LEfSe identified many key bacterial phylotypes, mainly at the genus and species levels, that could potentially distinguish children with T1 DM from healthy controls (Figure 3 A and B). A representative cladogram demonstrated dysbiosis of the T1 DM-associated fecal microbiota among children with T1 DM.
Specifically, no one phylum was observed to differ significantly between children with T1 DM and healthy controls. However, the dysbiotic indicator, the Firmicutes/Bacteroidetes (F/B) ratio, was markedly increased in children with T1 DM (Supplementary Figure 1), which suggested that fecal microbial dysbiosis occurred in patients with T1 DM. At the order level, we found that Erysipelotrichales, Enterobacteriales,and Coriobacteriales were enriched in children with T1 DM, while Selenomonadales was markedly reduced (P< 0 .05 ; Figure 3 C). At the family level, Lachnospiraceae,Erysipelotrichaceae, Enterobacteriaceae, and Coriobacteriaceae were significantly enriched in children with T1 DM (P < 0 .05 ; Figure 3 C).
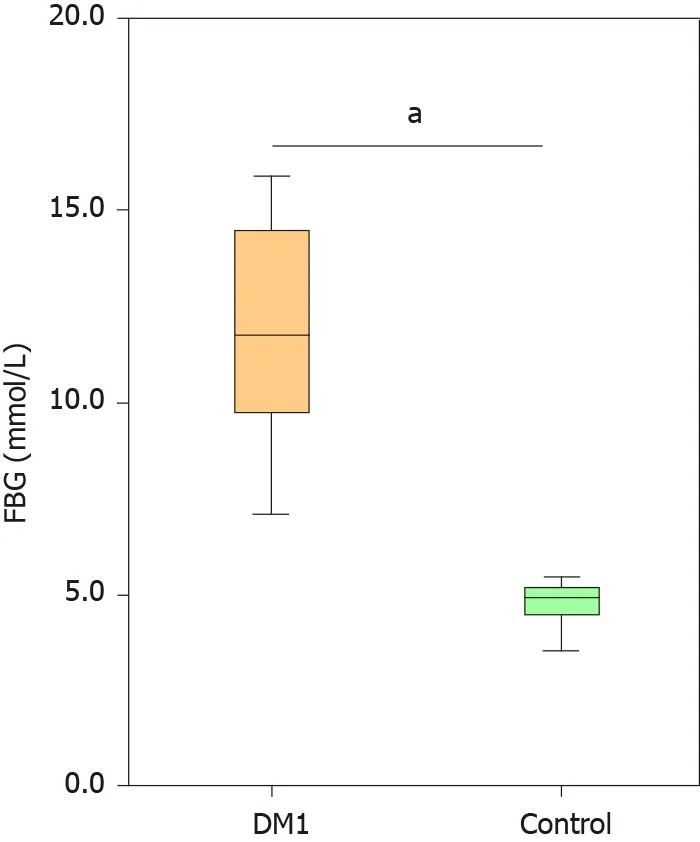
Figure 1 Comparison of the levels of fasting blood glucose between Chinese children with type 1 diabetes mellitus and healthy controls.Data are presented as the mean ± SD. aP < 0 .05 , compared with control group. FBG: Fasting blood glucose; DM1 : Type 1 diabetes mellitus.
At the genus level,Blautia,Anaerostipes, unclassified Lachnospiraceae, theEubacterium halliigroup, unclassified Peptostreptococcaceae,Dorea,Collinsella, andKlebsiellawere significantly enriched in children with T1 DM, whereasParabacteroides,Flavonifractor, and uncultured Ruminococcaceae were markedly reduced (P< 0 .05 ;Figure 3 C). At the species level, Anaerostipes hadrus, Ruminococcus sp._5 _1 _39 BFAA,Dorea longicatena, andCollinsella aerofacienswere enriched in children with T1 DM,while seven species, namely,Bacteroides vulgatusATCC8482 ,Bacteroides ovatus,Bacteroides xylanisolvens,Bacteroides dorei,Flavonifractor plautii,Parabacteroides merdae,andParabacteroides distasonisATCC8503 were markedly reduced (P < 0 .05 ; Figure 3 C).Figure 4 shows a heatmap of bacterial genera in children with T1 DM and healthy controls, presenting the relative percentages of most genera identified in each sample.
The overall structure of the T1 DM-associated fecal microbiota was the result of dynamic interactions between community members. The SparCC algorithm with false discovery rate adjustment was employed to generate correlation-based microbial interaction networks based on the relative abundance of OTUs between the two groups (Figure 5). We found a more complex network of interactions in healthy controls than in children with T1 DM. More positive and negative correlations among bacteria were found in the healthy controls. Based on our present findings, dysbiosis of the T1 DM-associated fecal microbiota was observed in children with T1 DM.
A fecal microbiota-based signature discriminates patients with T1 DM from healthy controls
As mentioned above, several taxa were identified as key functional differentially abundant bacteria. These differentially abundant taxa, includingBacteroides vulgatusATCC8482 ,Bacteroides ovatus,Bacteroides xylanisolvens, andFlavonifractor plautii, were negatively correlated with FBG, whileBlautia,Eubacterium halliigroup,Anaerostipes hadrus, andDorea longicatenawere positively correlated with FBG (P< 0 .05 ; Figure 6 ).These correlation analyses indicated that key T1 DM-associated functional bacteria actively participated in the regulation of glycemic levels in children.
We evaluated the potential value of the key functional differentially abundant taxa as biomarkers, includingBacteroides vulgatusATCC8482 ,Bacteroides ovatus, theEubacterium halliigroup, andAnaerostipes hadrus. First, using only one of the differential bacteria as a predictor, we generated the receiver operating characteristic curves,with the area under the curve (AUC) ranging from 0 .294 to 0 .690 (Figure 7 A).Multivariable stepwise logistic regression analysis was then performed to evaluate the list of T1 DM-associated taxa, to distinguish T1 DM patients from healthy controls(Figure 7 B). We found that a combination of four taxa, includingBacteroides vulgatusATCC8482 ,Bacteroides ovatus, theEubacterium halliigroup, andAnaerostipes hadrus,could significantly improve the predictive performance (AUC = 0 .830 ). Based on our present findings, these key differentially abundant taxa could be used as potential biomarkers for discriminating between T1 DM patients and healthy controls.
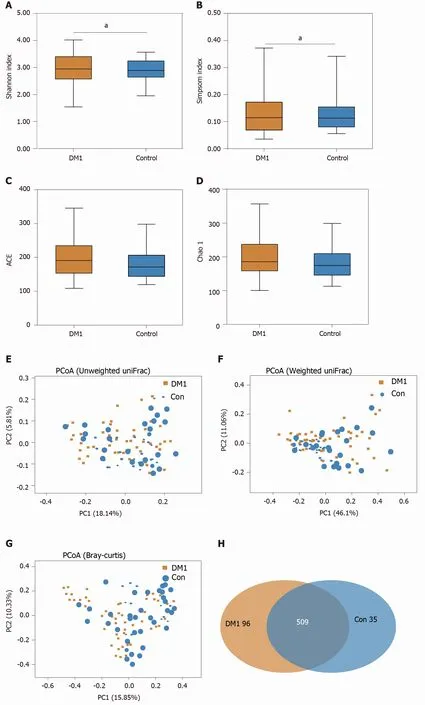
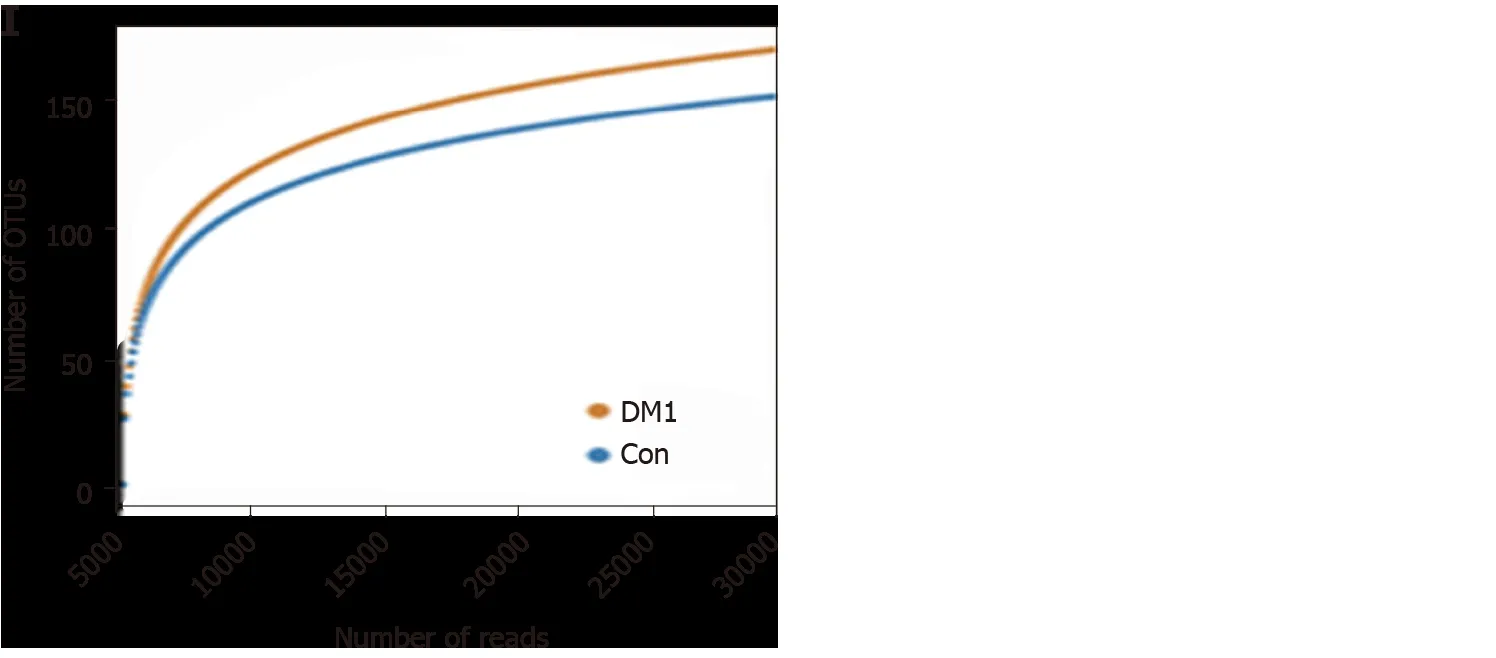
Figure 2 Altered bacterial diversity and richness of the fecal microbiota in Chinese children with type 1 diabetes mellitus. A-D: The diversity indices, such as Shannon (A), and Simpson (B), and the richness indices, such as the abundance-based coverage estimator (C), and Chao1 (D), were used to evaluate the overall structure of the fecal microbiota in patients with type 1 diabetes mellitus (T1 DM) and healthy controls. Data are presented as the mean ± SD.Unpaired t-tests (two-tailed) were used to analyze variation between the two groups; E-G: Principal coordinate analysis plots of individual fecal microbiota based on unweighted (E) and weighted (F) UniFrac distance, and Bray–Curtis dissimilarity (G) in patients with T1 DM and healthy controls. Each symbol represents a single sample; H: Venn diagram illustrating the overlap of operational taxonomic units (OTUs) in T1 DM-associated fecal microbiota between the two groups; I: Rarefaction curves usedtoestimate therichness (ata97 %levelof similarity)of T1 DM-associatedfecalmicrobiota betweenthe two groups.The vertical axisshows the expected numberofOTUsafter samplingthe numberoftagsor sequences shown onthehorizontalaxis. aP < 0 .05 .OTUs: Operationaltaxonomic units;ACE: Abundancebased coverage estimator; PCoA: Principal coordinate analysis; Con: Control; DM1 : Type 1 diabetes mellitus.
T1 DM-associated microbial functional alterations
To study the functional and metabolic changes in microbial communities between patients with T1 DM and controls, we inferred the metagenomes from the 16 S rRNA data and analyzed the functional potential of the fecal microbiota using PiCRUSt software, based on closed-reference OTU picking. We compared 64 Kyoto Encyclopedia of Genes and Genome (KEGG) pathways at level 2, and identified five KEGG categories with significantly differential abundances between children with T1 DM and healthy controls. We found that glycan biosynthesis and metabolism and lipid metabolism were significantly reduced in children with T1 DM (P < 0 .05 ;Figure 8), which suggests that they might play crucial roles in the development of T1 DM.
Specifically, nine pathways at level 3, including transcription factors, the phosphotransferase system, methane metabolism, and peptidoglycan biosynthesis, were significantly increased in the T1 DM-associated fecal microbiota. Furthermore, 18 other pathways, including glycan degradation, glycosaminoglycan degradation, and insulin signaling pathway, were markedly reduced in the T1 DM-associated microbiota.Together, these functional alterations in the fecal microbiota, especially that in glycan metabolism, were likely associated with the pathogenesis and development of T1 DM.
DISCUSSION
As a chronic autoimmune disease, T1 DM is affected by genetic and non-genetic factors. With the advent of high-throughput sequencing technology, numerous studies have found that non-genetic factors, such as an optimal balance of the gut microbiota,play vital roles in regulating the host immune system and preventing the development of T1 DM. A recent study demonstrated that a dysbiotic gut microbiota limits the effects of therapy in T1 DM, while depletion of gut microbiota resistance enhances stem cell therapy in T1 DM[17 ]. Our present T1 DM-associated gut microbiota analysis excluded the influence of physiological factors such as the age, gender, and race of the enrolled participants. Most of the children with T1 DM were newly diagnosed cases and were treated with only insulin. Without any additional treatment options, our present microbiota analysis could determine the actual correlations and roles of gut bacteria in the development of T1 DM.
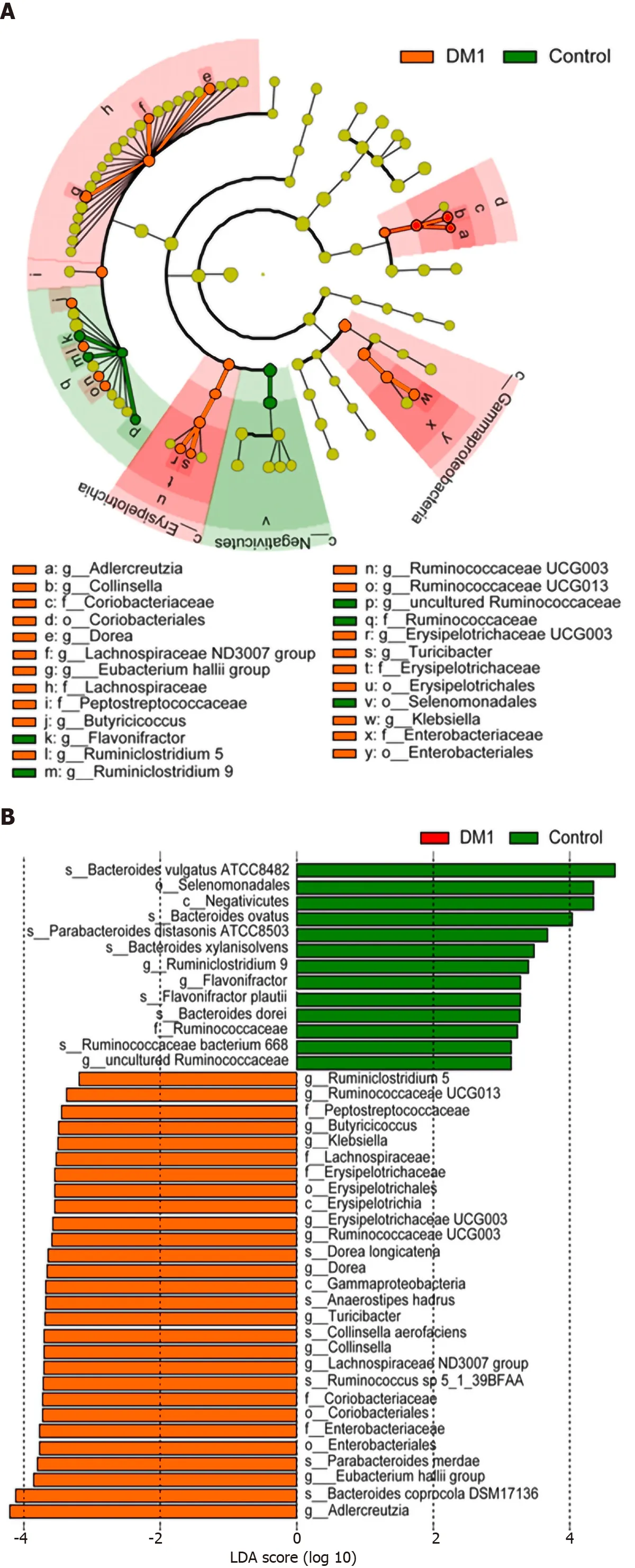
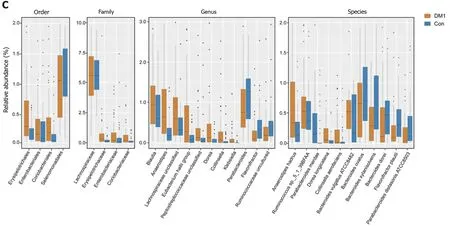
Figure 3 Differential bacterial taxa between Chinese children with type 1 diabetes mellitus and healthy controls. The linear discriminant analysis effect size identifies the taxa with the greatest differences in abundance between children with type 1 diabetes mellitus (T1 DM) and healthy controls. A and B: Only the taxa meeting a significant linear discriminant analysis threshold value of > 2 are shown; C: Comparisons of the relative abundance of abundant bacterial taxa at the levels of the order, family, genus, and species. Data are presented as the mean ± SD. Mann–Whitney U-tests were used to analyze variation between children with T1 DM and healthy controls. Con: Control; DM1 : Type 1 diabetes mellitus.
In the present study, the bacterial diversity of the T1 DM-associated fecal microbiota was significantly increased[6 ,8 ,10 ,18 ], which is inconsistent with the findings of previous studies. Several case-control studies have reported that the bacterial diversity of the gut microbiota is not significantly different in children with T1 DM[8 ,10 ,18 ].However, Leiva-Geaet al[6 ] found that T1 DM is associated with a significantly lower microbiota diversity[6 ]. Furthermore, de Goffau et al[5 ] indicated that the age of children may influence bacterial diversity, and older children with T1 DM tend to have a higher bacterial diversity[5]. In addition, differences in geographic location could influence the gut microbiota in early childhood, which might explain the disparity in bacterial diversity among different T1 DM-associated microbiota studies. As significant inter-personal variations were observed, our PCoA could not separate the children with T1 DM from healthy controls in these case-control studies. This indicates that βdiversity was similar between children with T1 DM and healthy controls. In contrast to microbiota shifts in other childhood diseases, such as antibiotic-associated diarrhea,our present α- and β-diversity analyses showed increased bacterial diversity in children with T1 DM, which could be used as a potential target dietary intervention in T1 DM.
Inconsistent with previous findings regarding bacterial diversity, our LEfSe analysis showed that several taxa could be used as biomarkers to discriminate T1 DM children from healthy controls. Consistent with previous human and animal studies,Bacteroidetes and Firmicutes were the two most predominant phyla, accounting for more than 93 % of the gut microbiota in children. Most of the differentially abundant taxa at the genus and species levels belonged to these two phyla. However, the dominant phyla showed no significant differences between the two groups. The composition of the gut microbiota at the phylum level varied with age, and a greater relative abundance of Bacteroidetes was observed among older individuals. Despite the absence of differentially abundant phyla, the F/B ratio was significantly higher in children with T1 DM. The F/B ratio is an indicator of gut dysbiosis, which is positively correlated with BMI[19 ]. Alterations in the F/B ratio may be important, as this ratio can influence efficiency in the processing of indigestible complex polysaccharides in the diet[20 ,21 ]. A previous study reported that the F/B ratio showed a significant decline in adults with type 2 diabetes mellitus (T2 DM)[22 ]. Although the change patterns are not always consistent in patients with diabetes, an altered F/B ratio demonstrates a dysbiotic gut microbiota compared with healthy controls.
Specifically, we found that several genera and species of the phyla Firmicutes and Bacteroidetes were significantly altered in the T1 DM-associated fecal microbiota.Interestingly, the co-network analysis indicated that interactions among altered bacterial species and abundant species play an important role in shaping the overall structure and composition of the T1 DM-associated fecal microbiota. We found a more complex network of interactions in healthy controls than in children with T1 DM, with more positive and negative correlations in healthy controls. These differentially abundant bacterial species played vital roles in regulating blood glucose in children.Bacteroides vulgatusATCC8482 , a highly abundant gram-negative obligate anaerobe,constitutes part of the core gut microbiota in healthy humans and is generally considered beneficial[23 ]. We found that the level of Bacteroides vulgatus ATCC8482 was significantly reduced in the T1 DM-associated fecal microbiota and negatively correlated with FBG. Leiva-Geaet al[6] also found that the prevalence ofBacteroides vulgatuswas significantly reduced in patients with T2 DM, which can be considered a gut microbiota signature associated with the development of T2 DM. Pedersen et al[24 ]identifiedBacteroides vulgatusas the main species driving the association between the biosynthesis of branched-chain amino acids (BCAAs) and insulin resistance,suggesting that it may directly impact host metabolism[24 ]. Similar to our present findings, Yoshidaet al[25 ] also revealed a significantly lower abundance ofBacteroides vulgatusin patients with coronary artery disease. Gavage with liveBacteroides vulgatuscan attenuate atherosclerotic lesion formation in atherosclerosis-prone mice. Such action can thereby markedly ameliorate endotoxemia, directly reduce gut microbial lipopolysaccharide (LPS) production, and effectively suppress proinflammatory immune responses. These studies suggest thatBacteroides vulgatusplays a beneficial role in regulating blood glucose and preventing the development of T1 DM.

Figure 5 Correlation strengths of the abundant fecal microbiota in Chinese children with type 1 diabetes mellitus and healthy controls.Correlation network of the abundant fecal microbiota in healthy controls and children with type 1 diabetes mellitus (T1 DM) is shown. Correlation coefficients were calculated with the sparse correlation for compositional data algorithm. The Cytoscape version 3 .4 .0 software was used for network construction. Red and blue lines represent positive and negative correlations, respectively. The correlation network became simpler in T1 DM. DM1 : Type 1 diabetes mellitus.
Another dominant species of the genusBacteroidesin the human gut microbiota,Bacteroides dorei, shares similar 16 S rRNA sequencing patterns withBacteroides vulgatus. Leonardet al[26 ] found that cesarean section delivery is associated with a reduced abundance of the beneficial species,Bacteroides vulgatusandBacteroides dorei[27 ]. Altered relative abundance ofBacteroides doreican significantly influence the composition of the gut microbiota, and this species can be considered a keystone species[27 ]. A previous study showed that increased abundance ofBacteroides doreileads to reduced gut microbial production of LPS, which improves immune function through mechanisms such as major histocompatibility complex production and T cell activation[25 ]. Anonye et al[28 ] also found thatClostridium difficilegrowth is significantly reduced in the presence ofBacteroides dorei[28 ]. However, a recent study on gestational diabetes identifiedBacteroides doreias a putative biomarker of impaired carbohydrate tolerance, and suggested that it played a role in the regulation of glucose tolerance in pregnant women[29 ]. However, our present correlation analysis found no significant association betweenBacteroides doreiand FBG. Inconsistent with our present alteration patterns, previous studies have found that higher levels ofBacteroides doreimay contribute to the onset of T1 DM, and may be potential monitoring and therapeutic microbial markers for T1 DM[30 ].Bacteroides ovatusis a common member of the human gut microbiota, with a broad capability to degrade complex glycans[31 ].It makes considerable contributions to the overall differences between T1 DM cases and controls. Consistent with our results, Giongoet al[14 ] found thatBacteroides ovatusaccounted for nearly 24 % of the total increase in the phylum Bacteroidetes among children with T1 DM[14 ] and for the first time established a causal relationship betweenBacteroides ovatusand metabolic homeostasis. Their findings demonstrated thatBacteroides ovatusmay be a potentially beneficial intestinal bacterial species.Similar toBacteroides vulgatus,Bacteroides ovatuscan also regulate BCAA metabolism,and alleviate metabolic syndrome. A recent study performed by Yanget al[32 ]demonstrated that specific strains ofBacteroides ovatusare capable of inducing high levels of mucosal immunoglobulin A (IgA) production in the large intestine, which can be used to modulate the host immune response[32 ]. In addition, as one of the active immunomodulators, oral gavage ofBacteroides ovatuscould significantly increase the efficacy of erlotinib and induce the expression of CXCL9 and interferon-gamma in a murine lung cancer model, which was positively correlated with treatment outcomes[33 ]. Another Bacteroides species with reduced levels in T1 DM,Bacteroides xylanisolvens,is a xylan-degrading bacterium isolated from human feces. Following a safety evaluation of aBacteroides xylanisolvensstrain (DSM 23964 ), a previous study reported its potential probiotic properties[34 ]. Consistent with a previous study on patients with atherosclerosis[35 ],Bacteroides xylanisolvensis reportedly an important contributor to folate transformations II and glycolysis III, and it is significantly more abundant in healthy controls than in patients with T1 DM. Qiao et al[36 ] also found thatBacteroides xylanisolvenscan alleviate nonalcoholic hepatic steatosis and provided evidence of the benefits of the gutBacteroides-folate-liver pathway[36 ].Bacteroides xylanisolvensis considered a probiotic bacterium that is positively correlated with antiinflammatory/tumor markers and negatively correlated with proinflammatory/tumor markers[37 ]. Sufficient evidence has supported and facilitated authorization of the use of heat-inactivatedBacteroides xylanisolvensin the European Union[38 ].Flavonifractor plautii, a Gram-positive anaerobic bacterium, is a member ofClostridiumcluster IV in the Ruminococcaceae family, and has been isolated worldwide from human feces. Our data showed a lower level ofFlavonifractor plautiiin children with T1 DM than in healthy controls. This finding suggests thatFlavonifractor plautiiplays a beneficial role in regulating the metabolism of blood glucose. Similar to our present findings, Borgoet al[39 ] found that Flavonifractor plautii is negatively correlated with BMI[39 ]. Kasaiet al[40 ] found that the fraction ofFlavonifractor plautiiis significantly lower in feces from obese subjects than in feces from non-obese[40 ]. Recently, Mikami et al[41 ] suggested that oral administration ofFlavonifractor plautiiprevents the accumulation of tumor necrosis factor-α-encoding transcripts in the adipose tissue of obese mice, thereby suppressing adipose tissue-associated chronic inflammation[41 ]. In addition, their group also found thatFlavonifractor plautiialleviates antigen-induced Th2 immune responses, and can be used as a potential anti-allergic probiotic[42 ].Flavonifractor plautiiabundance in fecal samples has now been proposed as a biomarker of health status[39 ].Parabacteroides distasonis, a core member of the gut microbiota in humans, is also reportedly a beneficial commensal gut microorganism in different pathophysiological models due to its anti-inflammatory and barrier restorative abilities. The abundance ofParabacteroides distasonisis relatively low in patients affected by obesity,nonalcoholic fatty liver disease (NAFLD), and multiple sclerosis[43 -45 ]. A recent study indicated thatParabacteroides distasonismodulates host metabolism and alleviates obesity and metabolic dysfunctionsviathe production of succinate and secondary bile acids[46 ]. Colonization of antibiotic-treated or germ-free mice with a singleParabacteroides distasonisstrain induced Treg differentiation[43 ]. The abundance of anotherParabacteroidesspecies,Parabacteroides merdae, was also reduced in children with T1 DM,indicating its beneficial role during T1 DM development. Wang et al[47 ] recently reported that enrichment ofParabacteroides merdaeis positively correlated with longevity[47 ]. These bacteria exhibit promising potential beneficial effects on human health in a strain-dependent manner. Thus, several strains could contribute to the development of chronic diseases.
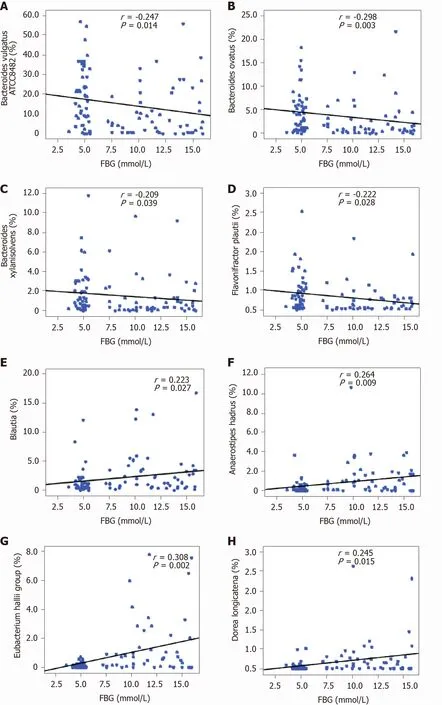
Figure 6 Correlation between key bacteria in type 1 diabetes mellitus-associated fecal microbiota and fasting blood glucose. The different taxa, including Bacteroides vulgatus ATCC8482 , Bacteroides ovatus, Bacteroides xylanisolvens, and Flavonifractor plautii, were negatively correlated with fasting blood glucose (FBG), while Blautia, the Eubacterium hallii group, Anaerostipes hadrus, and Dorea longicatena were positively correlated with FBG. Spearman’s rank correlation and probability were used to evaluate statistical significance. A: Bacteroides vulgatus ATCC8482 ; B: Bacteroides ovatus; C: Bacteroides xylanisolvens; D:Flavonifractor plautii; E: Blautia; F: Anaerostipes hadrus; G: Eubacterium hallii; H: Dorea longicatena. FBG: Fasting blood glucose.
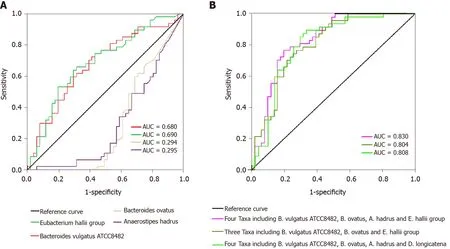
Figure 7 Receiver operating characteristic curves for different taxa, including Bacteroides vulgatus ATCC8482 , Bacteroides ovatus,Anaerostipes hadrus, and the Eubacterium hallii group, which were used either alone or in combination to discriminate between patients with type 1 diabetes mellitus and healthy controls. A: Alone; B: Combination. AUC: Area under the curve.
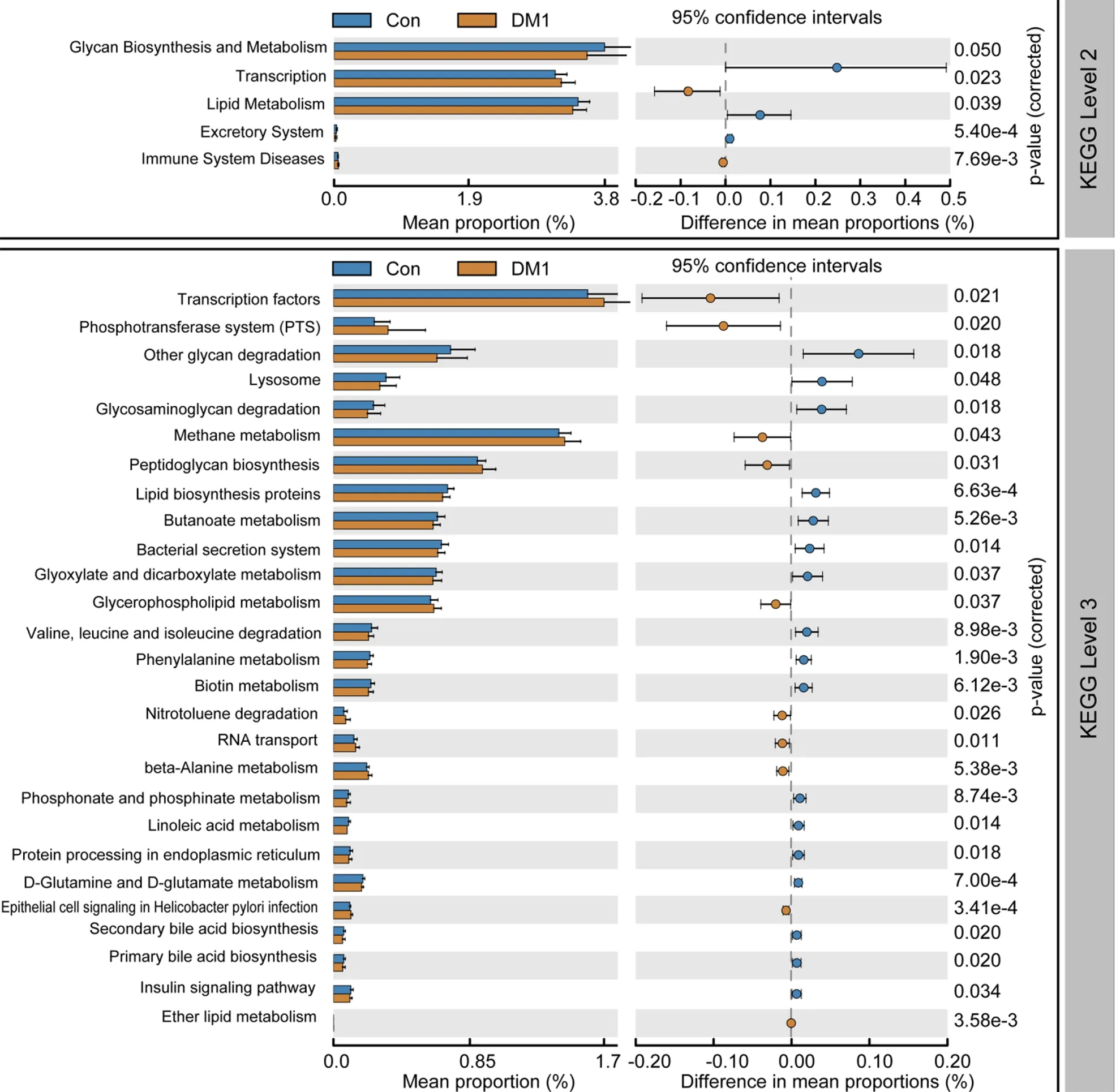
Figure 8 PiCRUSt-based fecal microbiome study among Chinese children with type 1 diabetes mellitus and healthy controls. Different bacterial functions were evaluated based on the two-sided Welch’s t-test. Comparisons between the two groups for each Kyoto Encyclopedia of Genes and Genome functional category (level 2 and level 3 ) are shown by percentage. The Benjamini–Hochberg method was used for multiple testing correction, based on the false discovery rate using the Statistical Analysis of Metagenomic Profiles software. KEGG: Kyoto Encyclopedia of Genes and Genome; Con: Control; DM1 : Type 1 diabetes mellitus.
The proportional abundances of four species, namely,Anaerostipes hadrus,Ruminococcussp. 5 _1 _39 BFAA,Dorea longicatena, andCollinsella aerofaciens, were significantly increased in children with T1 DM.Anaerostipes hadrus, one of the core species isolated from human feces, is able to produce large amounts of butyrate from both L-sorbose and xylitol and can consume acetate[48 ]. Zhang et al[49 ] found thatAnaerostipes hadruscan significantly aggravate colitis in dextran sulfate sodium-treated mice, but exerts no detrimental effects in healthy mice[49 ]. Recently, Zeevi et al[50 ] observed that several genomic structural variants ofAnaerostipes hadrusare negatively correlated with weight, waist circumference, median blood glucose levels, and BMI and positively correlated with HDL cholesterol levels[50 ]. The functions ofAnaerostipes hadrusstructural variants are not consistent with those of the wild type, which is strongly associated with lower metabolic risk.Ruminococcussp. 5 _1 _39 BFAA, a member of the genusBlautia, is positively associated with FBG. A previous study reported thatRuminococcussp. 5 _1 _39 BFAA is enriched among elderly patients with hypertension and reduced exercise capacity[51 ]. To date, no studies have extensively investigated the roles and mechanisms ofRuminococcussp. 5 _1 _39 BFAA.Blautiais associated with blood glucose regulation, lipid metabolism, and regulation of T cell differentiation[6 ,52 ]. However, increased proportions ofBlautiahave been reported in various diseases, such as IBS, NAFLD, and Crohn’s disease. A previous case-control study also found that the abundance ofBlautiais increased in children with T1 DM, and is positively correlated with HbA1 c, the number of T1 DM autoantibodies, and the titers of tyrosine phosphatase autoantibodies[52 ]. Consistent with the findings of Kosticet al[53 ], the present study demonstrated thatBlautiais positively correlated with the levels of FBG in children with T1 DM.Dorea longicatena, a new member ofClostridiumcluster XIVa in the Lachnospiraceae family, can produce acetate as a fermentation product. Mortaşet al[54 ] found thatDorea longicatenais significantly more abundant in individuals working the night shift[54 ]. Yang et al[55 ] found thatDoreais a biomarker of the risk for colorectal cancer[55 ]. Higher abundance of theDoreagenus is associated with increased intestinal permeability. In contrast with our present findings, Braheet al[56 ] reported thatDorea longicatenais negatively correlated with markers for insulin resistance, such as glucose and insulin, in obese female participants [56 ].Nevertheless, the increased abundance ofDorea longicatenain T1 DM could be actively involved in regulating host metabolism.Collinsella aerofaciens, one of the most abundant Actinobacteria in the gastrointestinal tract of healthy humans, shows increased abundance in the feces of patients with T1 DM; one of its subspecies is capable of butyrate production[57 ]. IncreasedCollinsellaabundance has been associated with both positive and negative health conditions, but there is no consensus on its health effects. Cohort studies have identified increases inCollinsellaabundance in the fecal microbiota of patients with T2 DM, atherosclerosis, and IBS[58 -60 ].Turnbaughet al[61 ] reported that the enrichment ofCollinsella aerofaciensis linked to BMI, with an increased prominence ofCollinsella aerofaciensin obese individuals than in lean twins and their mothers[61 ]. Another abundant bacterium in T1 DM, theEubacterium halliigroup (an anaerobic, Gram-positive, catalase-negative bacterium of the Lachnospiraceae family), is a metabolically versatile species that can contribute to intestinal butyrate and propionate formation[62 ,63 ]. We observed a positive correlation between theEubacterium halliigroup and FBG. Consistent with the present data, Yeet al[64 ] found that the abundance of theEubacterium halliigroup was significantly higher in patients with gestational diabetes mellitus (GDM) who failed to make lifestyle modifications for glycemic control, which is also positively correlated with FBG[64 ]. They also showed that theEubacterium halliigroup can be used to distinguish GDM patients and patients who failed glycemic control from healthy controls. As mentioned by Schwabet al[63 ], theEubacterium halliigroup may actively contribute to metabolic interactions[63 ]. However, Udayappan et al[65 ] observed that oral treatment with theEubacterium halliigroup can improve insulin sensitivity in db/db mice[65 ], which is inconsistent with the present findings. Our ROC analysis found that the bacteria mentioned above, such asBacteroides vulgatusATCC8482 ,
Bacteroides ovatus, theEubacterium halliigroup, andAnaerostipes hadrus, can be used as potential biomarkers to discriminate children with T1 DM from healthy controls. These bacteria in patients with T1 DM might contribute to alterations in microbial functions and actively participate in the development of T1 DM. Therefore, they could be used as novel targets for non-invasive diagnostic biomarkers and personalized treatment of T1 DM in the future.
There are several limitations of the present study. First, our case-control study only explored the characteristics of the T1 DM-associated fecal microbiota, whereas alterations in the fecal microbiota after successful treatment were not investigated.Second, the fecal microbial signature and corresponding metabolites, as well as the diagnostic model associated with T1 DM, still require further clinical studies with a larger sample size to validate the results. Third, the relatively weak correlations between key differential functional bacteria and FBG could not indicate an obvious primary or secondary relationship. More clinical indicators should be added into these correlation analyses in future studies. Fourth, culturomics should be used to identify T1 DM-associated bacteria. Further animal experiments could help to determine the cause-effect relationship between these bacteria and the pathogenesis of T1 DM.
CONCLUSION
In summary, the present study investigated the altered profiles of fecal microbiota in children with T1 DM. High-throughput sequencing identified the detailed composition and diversity of the T1 DM-associated fecal microbiota at a much deeper level. We found that bacterial diversity was significantly increased in the T1 DM-associated fecal microbiota. We also observed microbial compositional changes at different taxonomic levels. The proportions ofBacteroides vulgatusATCC8482 ,Bacteroides ovatus,Bacteroides xylanisolvens,Flavonifractor plautii,Anaerostipes hadrus, andDorea longicatenaat the species level andBlautiaand theEubacterium halliigroup at the genus level showed significant differences between children with T1 DM and healthy controls, and were markedly correlated with FBG. Furthermore,Bacteroides vulgatusATCC8482 andBacteroides ovatus, either alone or in combination, can be used as non-invasive diagnostic biomarkers to distinguish between patients with T1 DM and healthy controls. In addition, functional changes in the T1 DM-associated fecal microbiota suggest that the alterations are associated with the functions and metabolic activities of the microbiota, which might play vital roles in the pathogenesis and development of T1 DM. Thus, our comprehensive investigation of the T1 DM-associated fecal microbiota provides novel insights into the pathogenesis of T1 DM.
ARTICLE HIGHLIGHTS

ACKNOWLEDGEMENTS
The authors thank all of the participants who recruited patients in this study.
 World Journal of Gastroenterology2021年19期
World Journal of Gastroenterology2021年19期
- World Journal of Gastroenterology的其它文章
- Celiac Disease in Asia beyond the Middle East and Indian subcontinent: Epidemiological burden and diagnostic barriers
- Biomarkers in autoimmune pancreatitis and immunoglobulin G4-related disease
- Risk of hepatitis B virus reactivation in patients with autoimmune diseases undergoing non-tumor necrosis factor-targeted biologics
- Risk factors and prognostic value of acute severe lower gastrointestinal bleeding in Crohn’s disease
- Changes in the nutritional status of nine vitamins in patients with esophageal cancer during chemotherapy
- Effects of sepsis and its treatment measures on intestinal flora structure in critical care patients