
N,N-二烷基二甘酰胺酸类配体萃取Pr(Ⅲ)及在水溶液中的配位化学
2021-05-24 03:33马思齐杨志红朱礼洋杨素亮田国新
原子能科学技术 2021年5期
马思齐,张 燕,杨志红,朱礼洋,杨素亮,*,田国新,2,*
(1.中国原子能科学研究院 放射化学研究所,北京 102413;2.清华大学 核能与新能源技术研究院,北京 100084)
近年来研究发现,N,N-二(2-乙基己基)二甘酰胺酸(HDEHDGA,HL)对不同价态的镧系和锕系元素离子有一定的萃取能力。HL作为一种羧酸类多齿配体,在pH=1~4条件下可脱掉质子,以阴离子形式与金属离子结合形成中性萃合物,萃取金属离子时,有机相中无需引入伴阴离子,无中性萃取剂因离子对萃取而易产生三相的问题;在高酸度条件下能以中性萃取剂形式与金属离子形成阳离子萃合物,但硝酸根作为伴阴离子可与萃合物中配体羧基上的质子形成氢键,消除有机相中的离子对,大幅改善萃合物极性及溶解性。
张燕等[1-2]报道了HL从不同浓度硝酸溶液中萃取不同价态镧系和锕系元素离子,推测在pH=1~4至接近中性条件下,HL以典型羧酸类萃取剂的离子交换机理萃取金属离子;在强酸性条件下(硝酸浓度大于1 mol/L)以中性萃取剂的离子对机理萃取金属离子和伴阴离子;而在pH=1~4至强酸度(硝酸浓度大于1 mol/L)的中间区域可能以阴离子和中性配体共同存在的混合模式进行萃取。Sherif等[3-4]研究了pH=1时HL从氯离子介质中萃取三价镧系离子和Th(Ⅳ),发现HL/煤油体系对镧系元素具有良好的萃取能力和萃取容量。当有机相中萃取剂大幅过量时,与三价镧系离子基本形成以LnL3为核心的萃合物,且对轻镧系离子具有良好的分离性能。在水溶性小分子同系物N,N-二甲基二甘酰胺酸(HDMDGA, HL′)与镧系离子的配位反应研究中发现,小分子同系物与三价镧系离子配合物的稳定常数之比与HL分离三价镧系离子之间的分离因子有线性关系,且同样具有LnL3型核心萃合物的内层配位几何构型,与水溶液中LnL′3和固体化合物中LnL′3的配位几何构型相似。袁小兰等[5]研究了硝酸浓度为0.37 mol/L时HL对Dy(Ⅲ)的萃取,发现参与萃取反应的萃取剂分子数接近6,推测萃合物中萃取剂可能以二聚体形式存在,但无法根据斜率分析法所得结果推测出合理的配位模式。
为进一步揭示萃取剂与金属离子浓度比不同条件下HL萃取镧系和锕系元素离子的萃取机理,本文拟在以前研究的基础上,结合以HL为萃取剂的萃取过程和以HL′为配体的水溶液中的配位化学(HL具有较长碳链,不溶于水,HL′与HL有相同的官能团,HL′碳链更短,具有良好水溶性,便于将水溶液体系和萃取体系相互关联验证),着重研究HL从低pH值区域内的硝酸盐体系中萃取Pr(Ⅲ)的萃合物组成和结构。
1 实验
1.1 主要试剂与仪器
高氯酸钠、高氯酸镨六水合物(Ⅲ)、硝酸镨(Ⅲ)六水合物、1,3二异丙苯、、二甲酚橙、六次甲基四胺,分析纯,国药集团化学试剂有限公司;二甲胺水溶液(33%),化学纯,国药集团化学试剂有限公司;氢氧化钠标准溶液(1.023 mol/L)、高氯酸标准溶液(1.023 mol/L),美国福禄克(Fluke)公司;EDTA标准溶液,由乙二胺四乙酸二钠标准物质(粉剂摩尔标准,北京试剂公司)溶于去离子水定容制得。HL、HL′,实验室合成,合成路径参照文献[6-7],分子结构示于图1。准确称量一定量的HL′产品溶于去离子水中,采用酸碱滴定法确定产品为水合HL′,相对分子质量为181.2,纯度>99%(1H NMR分析);HL产品,经核磁、质谱、红外、元素分析等表征确认,纯度>99%。
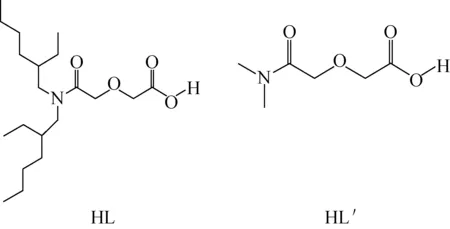
图1 HL和HL′分子结构
907-1 Titrando自动电位滴定仪,瑞士Metrohm公司,配备Pt 1000型温度补偿电极,电极填充液为1 mol/L NaCl溶液;Lambda950分光光度计,美国PerkinElmer公司;In Via激光显微拉曼光谱仪,英国Renishaw公司;BT125D分析天平(万/十万分之一),德国Sartorius公司;SHZ-D(Ⅱ)型循环水浴箱,巩义市予华仪器有限责任公司;ZWY-2102C恒温空气摇床,上海智诚分析仪器制造公司;Smart APEX Ⅱ CCD衍射仪,德国Bruker公司。
1.2 Ln(Ⅲ)盐溶液及HL和HL′溶液配制
1) 以Pr(Ⅲ)六水合高氯酸盐和六水合硝酸盐配制金属离子溶液, 采用EDTA络合滴定法确定金属离子浓度。
2) 准确称取适量HL′粉末定量溶解,室温下定容配制成0.3 mol/L HL′-0.7 mol/L NaClO4溶液(中和HL′中的0.27 mol/L质子,电位滴定实验用)和0.3 mol/L HL′-0.7 mol/L NaClO4溶液(中和HL′中的0.295 mol/L质子,光谱滴定实验用)。
3) 准确称取适量HL于烧杯中,加入1,3-二异丙苯溶解并于25 ℃定容,混匀,制得1 mol/L HL-1,3二异丙苯储备液。
1.3 HL′对Pr(Ⅲ)的萃取
为确定萃合物种类及组成,萃取实验分别在萃取剂浓度与金属离子浓度比不同的条件下进行。具体操作为:在离心管中加入1 mL有机相和对应量的NaOH溶液摇匀至乳白色,补足相应量的Pr(NO3)3溶液,水相与有机相相比为1∶1。恒温摇床控温至(25.0±0.1) ℃,机械振荡30 min,离心分相,分别取出水相、有机相,并通过分光光度法采集有机相的吸收光谱。
采用拉曼光谱法测定萃取后水相硝酸根浓度。通过标准曲线法计算萃取后水相的硝酸根浓度,采用差减法计算萃入有机相的硝酸根浓度[8]。
1.4 L′-与Pr(Ⅲ)配合物的稳定常数测定
以L′-与Pr(Ⅲ)为研究对象,确定L′-与Pr(Ⅲ)形成配合物的种类并获得配合物稳定常数,为获得准确数据并确定不同研究体系带来的影响,以两种方法分别测定配合物稳定常数。
1) 电位滴定法
Ln(Ⅲ)与L′-配合物的稳定常数以及HL′的质子化常数由电位滴定法获得。电位滴定仪参数见文献[9]。滴定前通过标准HClO4和NaOH溶液酸碱滴定校正电极。实验在Ar气氛下进行,以免CO2引入滴定杯对实验造成干扰;循环水浴温度控制在(25.0±0.1) ℃。每次滴定获取60~80个点,采用Hypquad2008软件计算配合物的稳定常数[10]。
2) 光谱滴定法
光谱滴定实验在10 mm×10 mm石英比色杯中进行,滴定前体积V0=2.0 mL、浓度c0(Pr(ClO4)3)=0.1 mol/L,控制离子强度为1 mol/L NaClO4。每次滴加0.1 mL 0.3 mol/L HL′-0.7 mol/L NaClO4溶液,充分摇匀(1~2 min,动力学表明配合反应可在30 s内完成),之后采用lambda950分光光度计测定其吸收光谱,Pr(Ⅲ)的光谱测量扫描范围为400~650 nm、数据间隔0.5 nm、狭缝为2.0 nm。滴定及光谱采集过程通过循环水浴将温度控制在(25.0±0.1) ℃。采用Hypspec软件[10]计算配合物稳定常数,并解析摩尔吸光谱。
1.5 晶体培养
为确定金属离子与配体的配位模式,培养单晶并解析晶体结构以获得相应数据进行佐证。培养方法如下:称取适量HL′粉末溶于无水乙醇中,制成1 mol/L HL′-乙醇溶液,二甲胺水溶液用于中和配体中的质子。PrL′3单晶在含0.2 mol/L Pr(ClO4)3和0.8 mol/L HL′-乙醇溶液中通过缓慢挥发获得。在100(10) K下由Bruker Smart APEX Ⅱ CCD衍射仪测定单晶的衍射数据,使用石墨化的单色MoK α辐射(λ= 0.0710 73 nm),并采用SADABS程序[11]对经验吸收进行校正。晶体结构通过直接方法求解,并使用SHELXL-97程序包在F2上用全矩阵最小二乘法精炼,使用骑乘模型将所有氢原子固定[12]。
2 结果及讨论
2.1 Pr(Ⅲ)与L′-的配合物稳定常数
N,N-二甲基二甘酰胺酸类配体在pH=1~4范围内以典型羧酸类化合物的离子交换机理与金属离子配位,配位模型如下:
在初始溶液离子强度I0=1 mol/L NaClO4、初始体积V0=20.9 mL、初始酸度c0(H+)=1.32 mmol/L、初始金属离子浓度c0(Pr(Ⅲ))=9.10 mmol/L条件下,Pr(Ⅲ)与L′-配位电位滴定曲线示于图2,滴定溶液浓度为:c(L′)=0.303 2 mol/L、滴定剂酸度c(H+)=0.031 1 mol/L。由图2可见,随着滴定剂体积的增加,逐渐生成了PrL′2+、PrL′+2、PrL′33种配合物,根据Hypquad软件[10]解得的L′-与Pr(Ⅲ)的配合物稳定常数(β)列于表1。

图2 Pr(Ⅲ)与L′-配位电位滴定图

表1 Pr(Ⅲ)与L′配合物的稳定常数
V0=2.0 mL、c0(Pr(Ⅲ))=0.1 mol/L、I0=1.0 mol/L NaClO4、c(L′)=0.302 2 mol/L、c(H+)=0.000 72 mol/L、滴定剂滴加量1.9 mL条件下的吸收光谱示于图3a,采用Hypspec软件解析,所得Pr(Ⅲ)与L′-的配合物的摩尔吸收光谱示于图3b。配合物稳定常数列于表1。
由图3a可见,随着滴定剂体积的增加, Pr(Ⅲ)在445、469、482 nm附近的3个特征吸收峰持续红移,且强度稍有减弱。由图3b可知:滴定过程中逐渐生成PrL′2+、PrL′+2、PrL′33种配合物;随着配体数的增加,3种配合物的特征吸收峰略有红移和减弱,且PrL′3配合物在590 nm处为双峰。

a——滴定过程中的光谱;b——解卷积的摩尔吸光谱
由表1看出,电位滴定和光谱滴定两种方法所得配合物稳定常数一致,配合物稳定常数数据可靠;与氯化物体系所得稳定常数略有差异,说明不同实验体系对配合物稳定常数有一定影响。
2.2 PrL′3晶体结构及配位模式
单晶X射线衍射法测得PrL′3单晶属三斜晶系,P1空间群,晶体参数为:a=0.997 (3) nm,b=1.033 (4) nm,c=1.985 (7) nm;α=94.999 (3)°,β=100.749 (3)°,γ=117.074 (4)°;V=1.753 (12) nm3,Z=2,ρcal=2.088 g/cm3,μ=1.632 mm-1,F(000)=908.0,配合物的晶体结构如图4所示。在PrL′3晶体中,Pr(Ⅲ)与L′-以三齿配体模式配位,在PrL′3配合物的内界中,以Pr(Ⅲ)为中心原子,Pr(Ⅲ)与L′-中的3个氧原子配位,形成9配位晶体,具有扭曲的三帽三棱柱几何结构。配体中参与配位的3个醚氧原子覆盖在3个棱镜的面上。该配合物没有氢键受体、供体或π键作用,结构几乎是孤立的,与LaL′3和NdL′[3]3的晶体结构相似。
2.3 HL对Pr(NO3)3的萃取
水相萃余的Pr(Ⅲ)浓度通过EDTA络合滴定测定,采用差减法计算有机相的Pr(Ⅲ)浓度。对采集到的有机相的吸收光谱进行处理,以各波长处吸光度A与有机相中Pr(Ⅲ)浓度的比值为纵坐标、以波长为横坐标作图,获得有机相的摩尔吸收光谱。
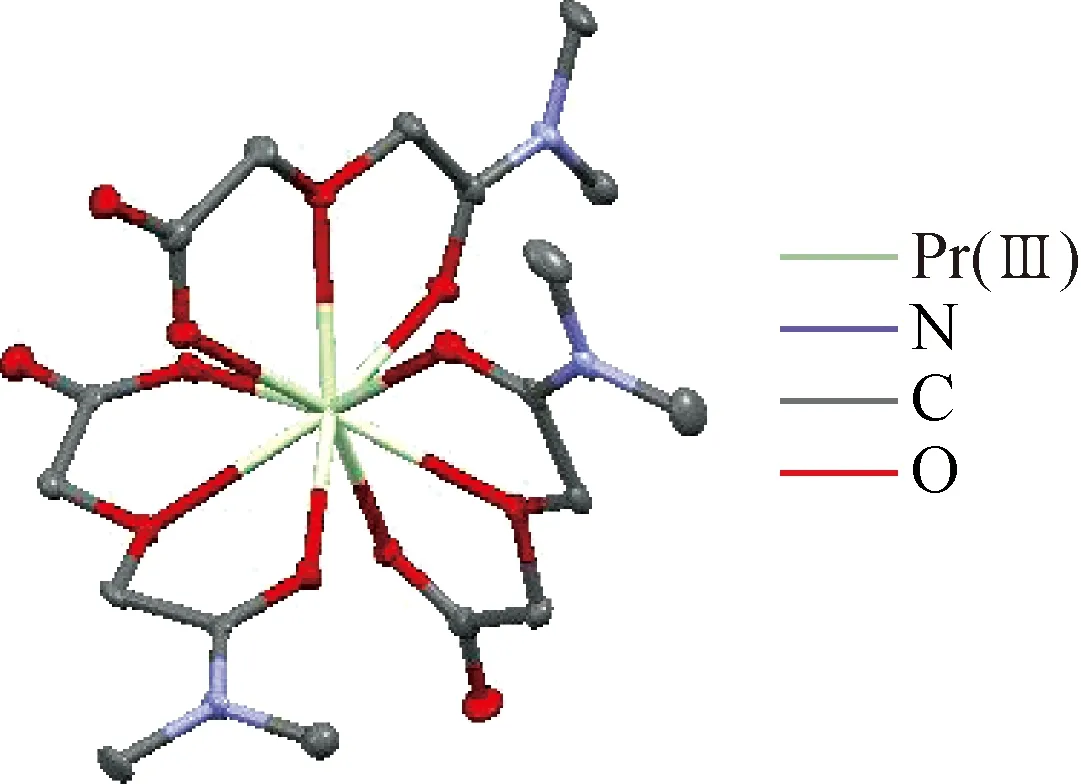
图4 沿a*轴具有30%椭球率的配合物PrL′3的晶体结构
水相硝酸根浓度由拉曼光谱法获得,In Via激光显微拉曼光谱仪参数如下:激发波长532 nm、光谱分辨率1 cm-1、激光功率20 mW。测定前用标准硅片校正峰位。以水作为内标,以系列已知样品中的硝酸根的摩尔浓度对硝酸根峰强度与水峰强度之比作图,获得标准曲线。

不同编号有机相萃取Pr(Ⅲ)后以Pr(Ⅲ)浓度归一化所得吸收光谱如图5所示。可看出起始有机相中萃取剂浓度小于水相中Pr(Ⅲ)浓度的2倍时,有机相中形成的萃合物基本都是组成为PrL2NO3的物种,吸收光谱也基本相同。而当起始有机相中萃取剂浓度大于水相中Pr(Ⅲ)浓度的4倍时,有机相中形成的萃合物基本都是组成为PrL3的物种,吸收光谱也基本相同。
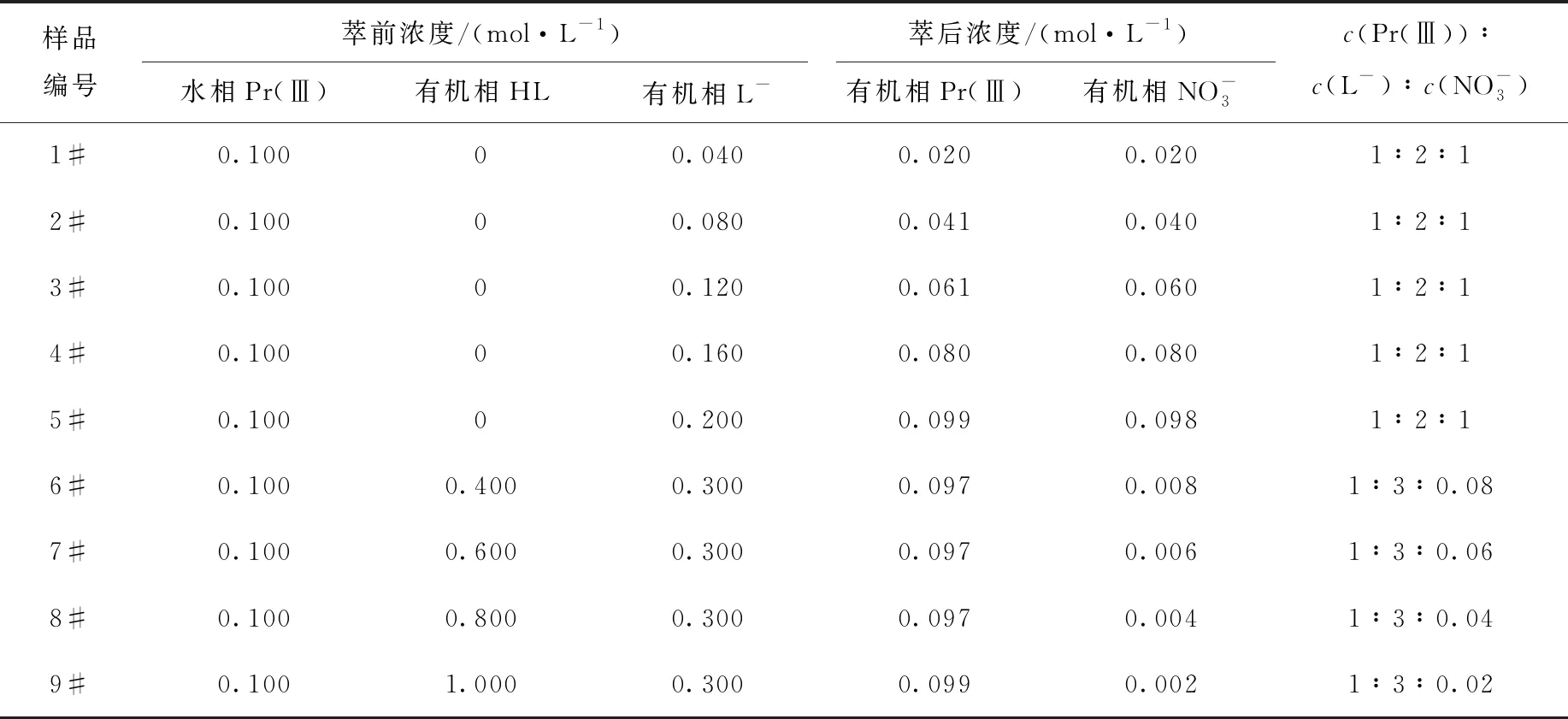
表2 萃合物制备条件及组成
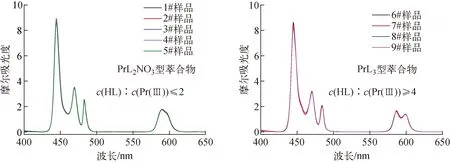
图5 HL萃取Pr(NO3)3后萃合物吸收光谱
与传统一元羧酸类[13-15]萃取剂萃取Pr(Ⅲ)时萃取剂与金属离子形成1∶3型萃合物不同的是,HL萃取Pr(Ⅲ)生成萃合物与萃取剂及Pr(Ⅲ)的相对含量有关,当萃取剂浓度与金属离子浓度比小于等于2时,主要生成PrL2NO3型萃合物,当萃取剂浓度与金属离子浓度比大于等于4时萃合物以PrL3为主。
2.4 固体光谱、水溶液光谱、萃取光谱相互关联
为确定HL与Pr(Ⅲ)的萃合物结构,将PrL′3的晶体漫反射光谱、光谱滴定解析得到的PrL′3和PrL′2+配合物的吸收光谱,与PrL2NO3及PrL3型萃合物的吸收光谱进行比较,结果示于图6。由图6a可见,PrL′3的晶体漫反射光谱、水溶液中PrL′3配合物的吸收光谱及PrL3型萃合物的吸收光谱的吸收谱带位置和形状非常相似,表明HL在萃取体系中与Pr(Ⅲ)的萃取模式与水溶液和固体PrL′3的配位几何模式一致,HL萃取Pr(Ⅲ)时形成的PrL3型萃合物中,L-以三齿配体的形式与Pr(Ⅲ)配位,形成与PrL′3晶体中类似的九配位结构。由图6b可见,水溶液中PrL′2配合物的吸收光谱及PrL2NO3型萃合物的吸收光谱在600 nm处峰形不同,推测PrL2NO3萃合物中硝酸根可能在内层参与配位。
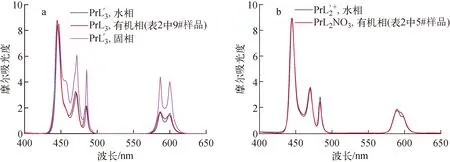
a——PrL3(有机相)、PrL′3(水相)的吸收光谱以及PrL′3(固相)的漫反射光谱 b——PrL′+2 (水相)与PrL2NO3(有机相)的吸收光谱
3 结论
在水溶液体系下通过光谱滴定和电位滴定研究了N,N-二甲基二甘酰胺酸离子L′-与Pr(Ⅲ)的配位,确定了PrL′2+、PrL′+2、PrL′33种配合物的生成,计算得到配合物的lgβ分别为4.62、7.98、10.12。用单晶X射线衍射技术测定了固体配合物PrL′3的晶体结构,确认PrL′3晶体中Pr(Ⅲ)是9配位,L′以三齿配体形式与Pr(Ⅲ)配位,形成扭曲的三帽三棱柱几何结构。以1,3-二异丙苯为溶剂,研究了HL对硝酸盐溶液中Pr(Ⅲ)的萃取。结果表明,脱质子的L-对Pr(Ⅲ)有较强的萃取能力,通常情况下形成PrL3萃合物,而当萃取剂浓度与金属离子浓度比较小(即脱质子的L-不足)时会形成PrL2NO3型萃合物。通过吸收光谱和拉曼光谱计算所得的有机相内硝酸根含量验证了萃合物为PrL2NO3。
比较PrL′3配合物在水溶液中的吸收光谱、固体漫反射光谱以及有机相中PrL3萃合物吸收光谱可知,HL萃取Pr(Ⅲ)的PrL3型萃合物中L-的配位模式与水溶液中及固体中PrL′3型配合物中L′-的配位模式一致,说明HL萃取Pr(Ⅲ)时L-以三齿配体形式与Pr(Ⅲ)配位,形成九配位结构。而PrL2NO3与PrL′+2的光谱差异表明,萃合物PrL2NO3的配位几何构型与水溶液中PrL′+2的配位几何构型不同,硝酸根可能直接参与了配位。
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
原子与分子物理学报(2020年5期)2020-03-17
当代陕西(2019年6期)2019-04-17
中国资源综合利用(2017年1期)2018-01-22
山东工业技术(2016年15期)2016-12-01
浙江大学学报(工学版)(2016年9期)2016-06-05
广东石油化工学院学报(2016年3期)2016-05-17
中国粮油学报(2016年5期)2016-01-23
应用化工(2014年8期)2014-08-08
郑州大学学报(理学版)(2014年3期)2014-03-01
