
Purification, characterization and hypoglycemic activity of glycoproteins obtained from pea (Pisum sativum L.)
2021-05-20 08:53GoyixinQinWuXuJunpingLiuLiynZhoGuitngChen
食品科学与人类健康(英文) 2021年3期
Goyixin Qin, Wu Xu, Junping Liu, Liyn Zho, Guitng Chen,*
a Department of Food Quality and Safety/National R&D Center for Chinese Herbal Medicine Processing, China Pharmaceutical University, Nanjing 210009, China
b College of Food Science and Technology, Nanjing Agricultural University, Nanjing 210095, China
Keywords:
Pea (Pisum sativum L.)
Glycoprotein
Structural characterization
Hypoglycemic activity
Simulated gastrointestinal digestion
ABSTRACT
This study aimed to isolate and characterize the structures of glycoproteins from peas and determine their hypoglycemic activity. The crude pea glycoproteins (PGP) were extracted by hot water and purified by diethylaminoethyl (DEAE)-Sepharose chromatography and Sephadex G-100 size-exclusion chromatography in sequence. Then three main fractions were obtained, namely PGP1, PGP2 and PGP3, with molecular weights of 897 615, 846 740 and 1 194 692 Da, respectively. The physical and chemical properties of the three fractions were evaluated and compared by Fourier transform infrared spectroscopy (FT-IR), nuclear magnetic resonance (NMR), scanning electron microscope (SEM), high performance liquid chromatography (HPLC)and other analytical techniques. The fraction PGP2 with the highest hypoglycemic activity, was screened using the Caco-2 monolayer cell model. It can inhibit the uptake of glucose in the small intestine, as well as the activities of maltase and sucrase. After simulated gastrointestinal digestion, PGP2 significantly enhanced the inhibitory effect of α-glucosidase, and slightly reduced the inhibitory ability of α-amylase. In summary, PGP2 possessed strong hypoglycemic activity after digestion. These results indicated that PGP2 has the potential to be developed into a functional food or natural medicine for the treatment of type 2 diabetes mellitus.
1. Introduction
Diabetes mellitus (DM) is a metabolic disorder caused by a variety of factors, characterized by chronic hyperglycemia, which is typically manifested in polydipsia, polyphagia, polyuria and weight loss [1]. DM is mainly divided into two types: the type 1 DM (T1DM),which is an autoimmune disease with insulin deficiency, and the type 2 DM (T2DM), which is mainly due to ineffective insulin action [2].T2DM accounts for almost 90% of the incidence of DM, which is a critical risk factor for many chronic diseases [3]. There are many diabetic patients in China, and the prevalence in adults over the age of 20 reached 9.7%. According to data released by the International Diabetes Federation in 2019, there are 463 million adults with diabetes worldwide [4]. Diabetes has become a public health problem that seriously endangers the health of global people.
At present, the mechanism of drugs for treating T2DM is mainly to increase peripheral blood insulin levels or reduce postprandial blood glucose levels or improve insulin resistance [5]. However, these drugs have some side effects, and they may cause drug dependence in patients. For example, sulfonylureas have been reported to cause hypoglycemia and weight gain [6]. Miglitol (an α-glucosidase inhibitor) can causes gastrointestinal side effects [7]. These natural products are stable in nature, having low toxicity and side effects.Besides, they have mild and long-lasting effects. While lowering blood sugar, these natural products also have the effects of lowering blood lipids, improving glucose tolerance and anti-inflammatory [8-10].
China is the third largest pea (Pisum sativum L.) producer in the world, with output accounting for one tenth of the world.Pea’s nutrition is comprehensive and balanced, it is rich in protein,carbohydrates and dietary fiber, while being extremely low in fat[11]. Ancient Chinese traditional medicine book Compendium of Materia Medica recorded that pea possesses the effect of treating diabetes. Recent studies have also revealed that peas have efficacy in treating diabetes. Studies by Tormo et al. [12] showed that feeding peas can significantly reduce blood glucose levels in diabetic rats, and Ramdath et al. [13] found that a high pea diet can effectively reduce the incidence of T2DM and low-density lipoprotein cholesterol.Ethanol extracts of peas showed potent anti-hyperglycemic effect in oral glucose tolerance test in diabetic mice [14]. Besides, the extracts of pea pod were useful to help manage diabetes and its associated hyperlipidemia, to reduce the risk of oxidative stress [15].
Glycoprotein is composed of two parts of sugar and protein connected by covalent bonds. It is an important biological macromolecule in the body and plays an important role in various life activities such as growth and development, information transmission,immune system, and nervous system [16]. The purpose of this study was to extract and separate different pea glycoproteins (PGP) from pea and compare their differences in structure and hypoglycemic activity. The component with the highest activity was selected through in vitro assays for further research, providing a theoretical basis for the future development of natural hypoglycemic drugs.
2. Materials and methods
2.1 Materials
The variety of dried peas is hard-pod green peas (‘Zhongwan-6’),which were produced and purchased from Baoding, Hebei Province,China. The color of pea cotyledons is green and the seeds are spherical. The content of protein and starch in the dried pea of this variety is 24.28% and 42.53%, respectively. Materials were identified by Prof. Liyan Zhao, and the voucher specimen (PSL20171027)was deposited at Department of Food Quality and Safety/National R&D Center for Chinese Herbal Medicine Processing, China Pharmaceutical University (Nanjing, Jinagsu, China). Dried peas were pulverized with a DFY-500 ultrafine grinder (Wenling Linda Machinery Co., Taizhou, Zhejiang, China) and sieved through a 0.15 mm mesh screen. Pea powder was stored in a sealed environment at-20 °C and used within one month.
DEAE-Sepharose (Fast Flow), α-glucosidase, α-amylase,pepsin and pancreatin were purchased from Shanghai Yuanye Bio-Technology Co. Ltd. (Shanghai, China). Sephadex G-100 was purchased from GE Healthcare Life Sciences (Pittsburgh, PA,USA). Acarbose was purchased from Beijing Solarbio Science& Technology Co. Ltd. (Beijing, China). Fetal bovine serum was purchased from Gibco (Grand Island, NY, USA). DMEM culture solution was purchased from HyClone (Logan, UT, USA). Caco-2 cells were purchased from Cell and Molecular Biology Experiment Platform of China Pharmaceutical University (Nanjing, Jiangsu,China). Transwell chambers was purchased from Corning Inc.(Corning, NY, USA).
2.2 Extraction and purification of PGP
The crude PGP were extracted as described in our previous report[17]. Crude PGP was dissolved in deionized water. After filtering with a 0.45 μm pore size membrane, a DEAE-Sepharose column(2.6 × 30 cm) was used for purification. The DEAE-Sepharose column was eluted by distilled water, followed by elution with gradient NaCl solutions (0.08, 0.16, 0.24 and 1 mol/L NaCl). Each fraction of 10 mL was collected at a flow rate of 5 mL/min. The protein content of each tube was measured at 280 nm, and total carbohydrate content was measured at 490 nm by phenol-sulfuric acid method. The same components were collected and concentrated by rotary vacuum evaporator, and then further purified with a column of Sephadex G-100. Each peak was collected and concentrated, and the fractions were freeze-dried after dialysis with the deionized water.
2.2.1 Protein content and polysaccharide content
The protein content was determined by the Bradford assay, using bovine serum albumin (BSA) as standard [18]. The polysaccharide content was determined by the phenol-sulfuric acid reaction, using glucose as standard [19].
2.2.2 Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
SDS-PAGE is generally used for glycoprotein analysis. A 10%separating gel and 5% stacking gel were used for SDS-PAGE, followed by Coomassie Blue staining and periodic acid-Schiff staining [20]. The operation process of periodic acid-Schiff staining was as follows. The 10% separating gel was treated with 10% trichloroacetic acid for 10 min to fix the sugar strips. After washing the gel with distilled water for 3 times, the gel was treated with 1% periodic acid for 30 min. Then the gel was washed with distilled water for 3 times to completely remove the periodic acid. The cleaned gel was stained with Schiff reagent for 30 min at room temperature in the dark, then washed with 0.5% sodium sulfite for 3 times, and stored in 7% acetic acid solution.
2.2.3 Monosaccharide composition determination
The analysis of monosaccharide composition was conducted according to the method described by Wu et al. [21] and Su et al.[22]. Three samples (2 mg) were hydrolyzed into monosaccharides component using 100 μL of 4 mol/L trifluoroacetic acid (TFA) at 100 °C for 2 h. After cooling to room temperature (24-26 °C), the hydrolyzed solution was co-evaporated with 200 μL of methanol to remove TFA, the procedure was repeated 3 times. The dried hydrolysate was dissolved in deionized water and filtered through a 0.22 μm membrane filter for subsequent derivatization.
The hydrolysates or monosaccharide standards (50 μL, dissolved in deionized water), 50 μL of 0.6 mol/L NaOH and 100 μL of 0.5 mol/L 1-phenyl-3-methyl-5-pyrazolone (PMP) (dissolved in methanol)were mixed and incubated at 70 °C for 2 h. After cooling to room temperature, the mixture was neutralized with 100 μL of 0.3 mol/L HCl, and then 1.5 mL of deionized water and 1.5 mL of chloroform were added, followed by vigorously shaking until homogeneous.The chloroform layer was discarded, and the extraction process was repeated 3 times. The aqueous layer was collected and filtered through a 0.22 μm membrane filter for high performance liquid chromatography (HPLC) analysis.
The PMP derivatives were analyzed by a Shimadzu HPLC-20A HPLC system equipped with a UV detector (SPD-M20A, Shimadzu Co., Kyoto, Japan) set at 245 nm. The separation of compounds was performed with an InertSustain C18column (4.6 mm × 150 mm, 5 μm,Shimadzu) at 30 °C. The PMP derivative solution was performed with a mixture of acetonitrile and 0.1 mol/L phosphate buffer saline (PBS)(pH 6.8) in a ratio of 17: 83 (V/V) at a flow rate of 1.0 mL/min. The injection volume was 20 μL.
2.2.4 Amino acid composition analysis
The method of Wang et al. [23] was used to analyze the amino acid composition with a Hitachi LA8080 Amino Acid Auto Analyzer(Hitachi Ltd., Tokyo, Japan). Accurate 100 mg of PGP1, PGP2 and PGP3 were weighed in a hydrolysis tube. After adding 15 mL of 6.0 mol/L hydrochloric acid and 3-4 drops of freshly distilled phenol,the hydrolysis tube was put in ice water to freeze for 3-5 min. Then the nitrogen- filled hydrolysis tube was hydrolyzed at 110 °C for 22 h.After cooling to room temperature, the hydrolysate was filtered and the volume was adjusted to 50 mL with deionized water. The filtrate(1 mL) was dried in a vacuum drying box at 50 °C and re-dissolved with 1 mL of pH 2.2 buffer for analysis.
2.2.5 Molecular weight determination
The molecular weight of PGP1, PGP2 and PGP3 were determined by HPLC (HPLC-20A, Shimadzu) equipped with TSK-GEL G4000PWXL column and an evaporative light scattering detector(ELSD-LT Ⅱ , Shimadzu). Column temperature was 30 °C. Three PGPs were eluted by deionized water at a flow rate of 0.5 mL/min. The molecular weight was estimated by reference to a calibration curve made from a set of Dextran standards (Mw10, 40, 70, 126 and 2 000 kDa).
2.2.6 Glycosylation type analysis
The glycosylation type was determined using the β-elimination reaction. Three samples were dissolved in 0.2 mol/L NaOH with 1.0 mol/L NaBH4for 3 h at 45 °C and then analyzed with a TU-1901 UV spectrophotometer (PERSEE General Instrument Co.,Beijing, China). The sample without sodium hydroxide treatment was used as a control [24].
2.2.7 Scanning electron microscope (SEM) analysis
SEM (S-3400N II, Hitachi) was applied to observe the surface characterization of PGP1, PGP2 and PGP3. Three samples were sputter-coated with gold layer before observation, and the images were collected at a voltage of 20.0 kV with magnification at 1 000 ×,3 000 ×, and 10 000 ×.
2.2.8 Fourier transform infrared spectroscopy (FT-IR)spectra analysis
FT-IR spectra analysis was carried out by using a FT-IR spectrometer (TENSOR 27, Bruker Co., Billerica, MA, USA). 1 mg of each sample (PGP1, PGP2 and PGP3) was mixed with 100 mg KBr powder, respectively, and then pressed into pellets for infrared spectral analysis within a range of 4000-400 cm-1.
2.2.91H and13C nuclear magnetic resonance (NMR) analysis
Twenty-five milligram of PGP1, PGP2 or PGP3 was dissolved in D2O in an NMR tube respectively. Spectra were recorded using Bruker AV-500 spectrometer (500 MHz, Bruker) for1H NMR and13C NMR at 25 °C.
2.3 In vitro hypoglycemic activity assays
2.3.1 α-Glucosidase inhibition assay
According to the experimental method of Qian et al. [25] and Chavez-Silva et al. [26], the dosage of the reagent was modified to determine the inhibitory activity of α-glucosidase. Acarbose was used as the positive control. The samples, enzymes and substrates used in the reaction were dissolved in 0.1 mol/L PBS(pH 6.8). After 3 PGP solutions/acarbose at different concentrations(200 μL) and α-glucosidase solution (0.2 U/mL, 40 μL) were incubated at 37 °C for 15 min, 40 μL of 5 mmol/L p-nitrophenyl-α-D-glucopyranoside (PNPG) solution was added. The reacting mixture was then incubated at 37 °C for 20 min and 200 μL of 1.0 mol/L Na2CO3was added to terminate the reaction. Before measuring the absorbance at 405 nm, the reaction system was diluted with 0.1 mol/L PBS to a suitable concentration. The control and blank referred to no α-glucopyranosidase or no glycoprotein sample in the system, respectively. The α-glucosidase inhibitory activity was expressed as inhibition rate. The inhibition rate was calculated using the following equation:

where AS, ACand ABare the absorbance of the sample, control and blank, respectively.
2.3.2 α-Amylase inhibition assay
The α-amylase inhibitory activity was performed according to the method of Rafique et al. [27] and Wu et al. [28] with minor modifications. Acarbose was used as the positive control. A 200 μL of α-amylase solution in 0.1 mol/L PBS (pH 6.9) with different concentration of 400 μL of 3 PGP samples/acarbose (0.1,0.2, 0.5, 1, 2 mg/mL) were incubated at 37 °C for 10 min. After preincubation, 1 mL of 1% starch solution was added into each test tube and incubated for 5 min at 37 °C. Then, 1 mL of dinitrosalicylic acid colorant was added to terminate the reaction, and the mixture was quickly cooled in a cold-water bath after incubating in boiling water bath for 5 min. The absorbance was measured at 540 nm after dilution of solution by adding 5 mL distilled water. The α-amylase inhibition rate was calculated as Equ (1).
2.3.3 Effect of three samples on cell proliferation
The safe concentration range of the 3 samples was determined by the methyl thiazolyl tetrazolium (MTT) assay [29]. Human colon cancer cells (Caco-2) on logarithmic growth phase were seeded in 96-well microplates at a density of 1 × 105cells/well. At the environment of 37 °C with 5% CO2saturation, the cells were incubated overnight to adhere. Then different concentrations (0,50, 100, 250 and 500 μg/mL) of PGP1, PGP2 and PGP3 were added. After incubated for 48 h, 20 μL of MTT solution (5 mg/mL)was added to each well and further incubated for 4 h. The supernatant was discarded and then added 150 μL of dimethyl sulfoxide. The absorbance was measured by a microplate reader(Elx800TM, BioTek Instruments Inc., Winooski, VT, USA) at a wavelength of 490 nm. The growth inhibition was calculated as the following equation:

where ASand ACare the absorbance of the treated and untreated cells, respectively.
The concentration range of samples in subsequent cell experiments was determined by the growth inhibition rate.
2.3.4 Establishment of Caco-2 monolayer cell model
The Caco-2 cells were subcultured every 4 days using a 0.125%ethylene diamine tetraacetic acid (EDTA) trypsin solution. Cell culture medium was changed every 2 days. The Caco-2 cells were trypsinized and seeded in 12-well transwells (0.4 μm) fitted in bicameral chambers at a density of 1 × 105cells/mL. On the 21st day after sowing, the integrity of the monolayer was checked by Hank’s balanced salt solution (HBSS) buffer containing fluorescent sodium salt, and fluorescent sodium salt was added to the top side (AP side)of the transwell cell. After incubation at 37 °C for 2 h, the solution on the bottom side (BL side) was pipetted for quantitative analysis,and the integrity of the cell monolayer model was evaluated by calculating the apparent permeability coefficient. The transepithelial electrical resistance (TEER) across the cell monolayer was measured using a Millicell ERS (Millipore, Eschborn, Germany) and the test requirement was satisfied when the value was greater than 500 Ω·cm2[30].
2.3.5 Inhibition of glucose uptake
After aspirating the culture medium from the transwell plate,the cells were washed with HBSS. Then added 400 and 750 μL of HBSS buffer to the AP side and the BL side, and incubated in a CO2incubator at 37 °C for 30 min. Then the HBSS buffer was aspirated.The experiment was divided into blank group, control group and sample group. In the blank group, 400 and 750 μL of HBSS buffer was added to the AP and BL sides of the transwell plate, respectively.In the control group, 400 μL of 25 mmol/L glucose solution was added to the AP side and 750 μL of HBSS buffer was added to the BL side of the transwell plate. In the sample group, 400 μL of 3 PGP components-glucose solution with different concentrations (25, 50 and 100 μg/mL) were added to the AP side, and 750 μL of HBSS buffer was added to the BL side. The reaction was incubated in a 37 °C incubator. When the reaction progressed to 30, 60, and 120 min,50 μL of the receiving solution was taken from the receiving chamber for testing, and the same volume of blank HBSS was immediately replenished on the BL side. The glucose oxidase assay kit (Shanghai Yuanye Bio-Technology Co. Ltd.) was used to detect the glucose content in the collected test solution. The content of glucose on the BL side of the sample group and the control group were used to calculate the inhibition rate of glucose in Caco-2 cells by different PGP components at different concentrations [31]. The inhibition rate was calculated as the following equation:
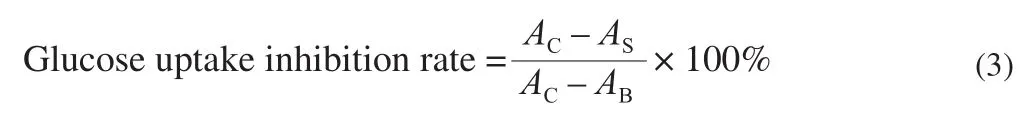
where AS, ACand ABare the absorbance of the sample, control and blank group, respectively.
2.3.6 Evaluation of ability to inhibit maltase and sucrase
The experimental operation process was same as the glucose uptake assay. The solution of the sample group was replaced with different concentrations of 3 PGP components-maltose solution and 3 PGP components-sucrase solution. The concentration of maltose solution and sucrase solution in the control group was both 28 mmol/L.
2.3.7 Simulated gastric and pancreatic digestion in vitro
The glycoprotein fraction with the best hypoglycemic activity screened by the above test was selected for in vitro simulated digestion. According to the con figuration method, 2.67 g of porcine pepsin (30 U/mg) and 0.2 g of NaCl was dissolved in 80 mL of distilled water, then HCl was added to adjust the pH of the solution to 1.2 and the volume was adjusted to 100 mL with distilled water to con figure an artificial gastric juice [32]. Artificial pancreatic juice was prepared by dissolving 0.04 g pancreatin (250 U/mg) in 50 mL of 50 mmol/L PBS with pH of 8.0 [33].
The screened glycoprotein fraction (100.0 mg) was dissolved in 50 mL gastric juice and incubated in a thermostatic shaker at 37 °C for 120 min, the reaction was immediately arrested by adding liquid nitrogen. About 25 mL of the reaction solution was taken for pancreatic digestion analysis. Pancreatic juice was added to the reaction solution at a ratio of 1:1 (V/V) and incubated under 37 °C for 120 min, finally the reaction was stopped by adding liquid nitrogen.The gastric digested sample was filtrate with distilled water to make the final concentration consistent with the pancreatic digested sample, then they were filtered with 0.45 μm filter membranes. The filtrate was utilized for the analysis of α-glucosidase and α-amylase inhibitory activity using methods already mentioned in sections 2.3.1 and 2.3.2.
2.4 Statistical analysis
All data are presented as mean ± standard deviation. Statistical analysis was performed with SPSS software (version 22.0 for Windows, SPSS Inc., Chicago, IL, USA). Statistical significance of the differences between groups was assessed by Student’s t-test.Differences between groups were considered statistically significant at P < 0.05.
3. Results and discussion
3.1 Purification of PGP
Crude PGP was separated on a DEAE-Sepharose column to obtain three main fractions (Fig. 1A). These fractions were further applied onto a Sephadex G100 column, only one fraction was obtained after purification, named as PGP1, PGP2 and PGP3 respectively(Fig. 1B-1D).
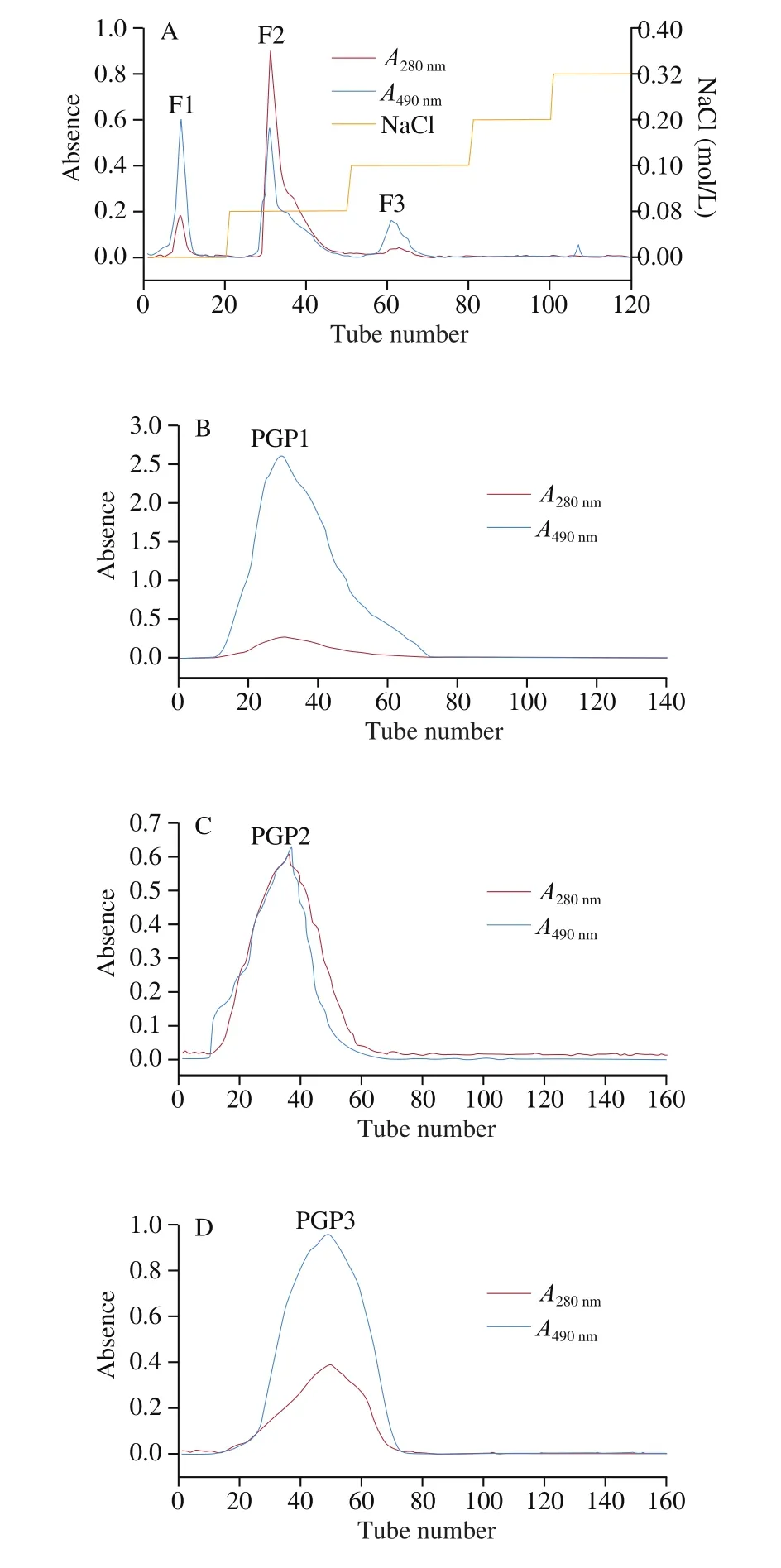
Fig. 1 Elution curve of PGP (A: Crude PGP was passed through a DEAE-Sepharose column to obtain three components; B, C, D: Three PGPs were further purified by sephadex G100).
3.1.1 Protein content and polysaccharide content
The protein content and polysaccharide content of the 3 glycoproteins were listed in Table 1. The purity of the 3 samples was all above 95%. Among the samples, PGP2 had the highest protein content, and PGP1 had the highest polysaccharide content.

Table 1Protein content and polysaccharide content.
3.1.2 SDS-PAGE analysis
The electrophoresis results of the three separated fractions were shown in the Fig. 2. The picture on the left showed the position of the protein bands of each fraction stained with Coomassie Brilliant Blue, and the picture on the right showed the position of the sugar bands stained with Schiff reagent. According to the staining results,the protein and sugar bands of each fraction appeared in the same position. Therefore, it can be determined that the three fractions were glycoproteins, rather than a mixture of proteins and polysaccharides.
Fig. 2 SDS-PAGE electropherogram (left: Coomassie blue stained band;right: Schiff reagent stained band).
3.1.3 Monosaccharide and amino acid composition
The monosaccharide and amino acid composition of the 3 glycoproteins were listed in Table 2 and Table 3. The chromatogram was shown in the Fig. 3. PGP1 was composed of mannose, ribose,rhamnose, glucose, galactose and arabinose. PGP2 was composed of mannose, ribose, galacturonic acid, glucose, galactose and arabinose. PGP3 was composed of mannose, rhamnose, glucuronic acid, galacturonic acid, glucose, galactose and arabinose. According to literature reported, polysaccharides with hypoglycemic activity basically contained several kinds of monosaccharides such as glucose,arabinose and galactose [34-37]. Among the 17 amino acids tested,PGP1 mainly contained aspartic acid, threonine, glutamic acid,leucine, tyrosine, phenylalanine and lysine. The contents of aspartic acid, glutamic acid, cystine, phenylalanine and lysine were high in PGP2. The protein part of PGP3 was mainly composed of aspartic acid, glutamic acid, tyrosine and phenylalanine.

Fig. 3 High-performance liquid chromatogram of 3 PGPs.

Table 2Monosaccharide composition of 3 PGPs.

Table 3The amino acid composition of 3 PGPs.
3.1.4 Molecular weight and glycosylation type
The molecular weights of PGP1, PGP2 and PGP3 were 897 615,846 740 and 1 194 692 Da, respectively. The molecular weights of PGP1 and PGP2 were relatively close and smaller than that of PGP3. The UV scanning spectra of 3 samples with and without alkali treatment were shown in Fig. 4. In contrast, 3 samples after alkali treatment had higher absorbance at 240 nm, which was indicative of theβ-elimination reaction. The results indicated that the proteins and carbohydrates in PGP1, PGP2 and PGP3 were connected byO-bonds.
3.1.5 SEM analysis
SEM images of three glycoproteins were shown in Fig. 5. The surface morphology of PGP1 and PGP2 was lumpy and stickshaped. Among them, PGP1 was mainly a stick-like structure, while PGP2 was mainly presented as a lumpy structure. And the structural morphology of PGP3 was mainly small spherical.
3.1.6 FT-IR spectra analysis
The FT-IR spectrum of 3 glycoproteins was shown in Fig. 6. The same characteristic absorption peaks appeared in the infrared spectra of these glycoproteins. The absorption peak in the range of 3 500-3 200 cm-1was the stretching vibration of O—H and N—H, indicating the existence of intermolecular and intramolecular hydrogen bonds[38]. The absorption band appearing in the range of 3 000-2 800 cm-1was caused by the stretching vibration of the C—H bond in the polysaccharide [39]. The peak at 1 647.16 cm-1was due to the carbonyl bond (C=O) stretching vibration of the amide group [40].A set of peaks from 1 400 cm-1to 1 200 cm-1were the variable-angle vibration of C—H bond, and the broad peak from 1 150 cm-1to 1 000 cm-1was the vibrational absorption of C—O bonds in sugar molecules. From the absorption peak near 1 075 cm-1, it can be inferred that the glycoside types of PGP1, PGP2 and PGP3 were mainly pyranose. PGP3’s unique absorption peak at 896.87 cm-1indicated that it contained β-glycosidic bonds.

Fig. 4 The UV scanning spectra of 3 PGPs.

Fig. 5 SEM images of 3 PGPs (A, PGP1; B, PGP2; C, PGP3).

Fig. 6 FT-IR spectrum of 3 PGPs.
3.1.7 NMR spectroscopy analysis
NMR spectra were used to analyze the chemical shifts of sugar residues that occur in the repeating units.1H and13C NMR spectra were shown in Fig. 7. The typical characteristics of chemical shifts of polysaccharides in1H NMR spectrum range from δ3 to 5 ppm, signals at around δ2.0 ppm represented protons of the N-acetyl group of the protein bonded residues [41]. The range from δ4.9 to 5.4 ppm was anomeric H-1 region, and the corresponding range of anomeric carbon was δ98 to 105 ppm [42]. It can be seen from the NMR spectrum that both PGP1 and PGP3 had peaks in this region, indicating the presence of α-glycosidic bonds in these two samples [43].

Fig. 7 1H (A) and 13C (B) NMR spectra of 3 PGPs.
3.2 In vitro hypoglycemic activity
3.2.1 Inhibition of α-glucosidase and α-amylase
α-Glucosidase secreted in the small intestine releases glucose from maltose and/or sucrose into blood resulting in post-prandial hyperglycemia and exacerbating noninsulin dependent or T2DM [44].In the process of optimizing the experimental conditions, we found that changes in substrate concentration had no effect on the degree of inhibition. Therefore, the inhibitory effects of the three PGPs on α-glucosidase and α-amylase might be reversible non-competitive inhibitions. The inhibition rates of α-glucosidase and α-amylase activity of the 3 PGPs were shown in Fig. 8 and Fig. 9, respectively.All three PGPs had inhibitory effects on α-glucosidase activity, and the strength of the inhibitory effects was concentration-dependent(Fig. 8). The inhibitory effects of PGP2 and PGP3 were similar with the change in concentration. As the increasing of concentration,the inhibitory effect gradually approached a stable value. However,when the concentration was less than 0.2 mg/mL, PGP1 had almost no inhibitory effect on α-glucosidase activity. When the concentration was higher than 0.2 mg/mL, the inhibitory effect of PGP1 increased rapidly with the increase of concentration, and the increase was faster than that of PGP2 and PGP3. In the positive control group, acarbose always had a strong inhibitory effect on α-glucosidase activity within the set concentration range, and the inhibition rate almost reached 100%.
From the results in the Fig. 9, 3 PGPs had inhibitory effects on α-amylase activity. The inhibitory effects of the three samples increased with increasing concentration, and finally reached a stable value. The inhibitory effects of acarbose was also positively correlated with the concentration, especially when the concentration was less than 0.5 mg/mL, the inhibition rate increased significantly with the increase of the concentration. The growth trend gradually slowed down after the concentration was higher than 0.5 mg/mL. However,the inhibitory effect of PGP1 was very weak. When the concentration was 2 mg/mL, its inhibitory rate on α-amylase was only about 2.73%,while the inhibition rates of PGP2 and PGP3 are 8.71% and 22.34%,respectively. At the same concentration, the inhibitory effect of PGP3 on α-amylase was better than that of PGP2.

Fig. 8 Inhibition rates of α-glucosidase activity.
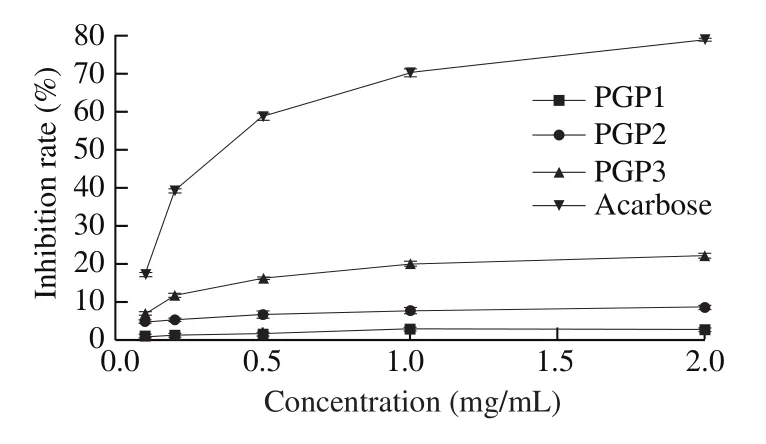
Fig. 9 Inhibition rates of α-amylase activity.
3.2.2 Cell proliferation
MTT assay was used to determine the proliferation toxicity of three glycoproteins on Caco-2 cells. As shown in Fig. 10, within the set concentration range, PGP3 basically had no toxic effect on Caco-2 cells, while PGP1 and PGP2 show strong cytotoxicity at 250 and 500 μg/mL. Therefore, the sample concentration in subsequent cell tests was in the range of 0-100 μg/mL.

Fig. 10 Effects of 3 PGPs at different concentrations on the growth of Caco-2 cells.
3.2.3 Evaluation of Caco-2 monolayer cell model
Cultured mature Caco-2 cells can form a tight unicellular layer tissue, whose structure and biochemical functions are similar with the human small intestinal epithelial cell barrier, and the cell monolayer differentiates into the villous surface A side and the basal surface B side. A side contains typical small intestinal microvilli hydrolase and various nutrient transporters, which can play the role of active transport substances [45,46]. The change of TEER value was shown in the Fig. 11.
On the 21st day of Caco-2 cell seeding, TEER values were all greater than 1 000 Ω·cm2, and the apparent permeability coefficient of sodium fluorescein was less than 0.5 × 10-6cm/s. It was proved that the cells in these wells had grown into a dense, complete monolayer.

Fig. 11 Change of TEER value of Caco-2 cell monolayer.
3.2.4 Inhibition of glucose uptake
The inhibition rate of glucose uptake was shown in Fig. 12. Under the conditions of the same sample concentration and the same reaction time, PGP2 had the best inhibitory effect on glucose uptake, followed by PGP3, and PGP1 had the worst inhibitory effect. With the increase of the reaction time, the inhibitory effects of the 3 samples showed a trend of increasing first and then weakening, and the effect reached the best when the reaction time was 1 h. The effect of the 3 samples increased with the increase of concentration. When the other conditions were the same, the effect of the 100 μg/mL sample was the best.

Fig. 12 Inhibition rate of glucose uptake. Significant (P < 0.05) differences are shown by data bearing different letters: in lowercase, between groups; in uppercase, within groups.
3.2.5 Ability to inhibit maltase and sucrase
Based on the results of Fig. 13, 3 glycoprotein samples at different concentrations all have a certain inhibitory effect on maltase activity. With the increase of the reaction time, the inhibitory effect of 3 glycoprotein samples on maltase also increased. Although the subsequent increase trend gradually slowed down, there was no trend of inhibitory effect decline within 2 h of the reaction, indicating that the inhibitory effect of 3 PGPs on maltase is relatively longlasting. It made maltose to be slowly broken down into glucose and enter the blood in the small intestine, preventing the glucose concentration in the blood from rising too quickly. Among 3 PGPs, PGP2 had the best inhibition on maltase, followed by PGP1 and PGP3 was the worst. Overall, 100 μg/mL of PGP2 had the best inhibitory effect on maltase activity.
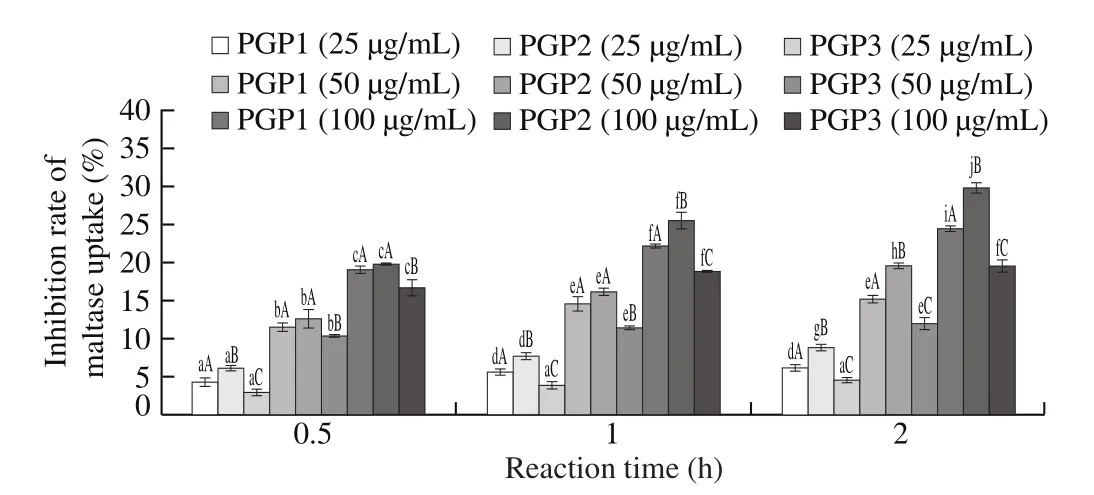
Fig. 13 Inhibition rate of maltase activity. Significant (P < 0.05) differences are shown by data bearing different letters: in lowercase, between groups; in uppercase, within groups.
From the results in the Fig. 14, all 3 glycoprotein fractions at different concentrations have a certain inhibitory effect on sucrase activity. Among the 3 samples, PGP2 had the best effect,PGP3 was the second, and PGP1 was the worst. The effects of the 3 glycoproteins were concentration-dependent. The higher the concentration was, the better the inhibitory effect on sucrase activity had. However, with the increase of the reaction time, the inhibitory effects of the 3 glycoproteins at different concentrations on the activity of sucrase were all weakened. When the reaction time reached 2 h, the inhibitory effect of the 3 glycoprotein samples on sucrase almost disappeared. It indicated that the 3 glycoprotein samples had a short inhibition time for sucrase activity.

Fig. 14 Inhibition rate of sucrase activity. Significant (P < 0.05) differences are shown by data bearing different letters: in lowercase, between groups; in uppercase, within groups.
3.2.6 Simulated gastric and pancreatic digestion in vitro
The results in Fig. 15 showed that the digested product of PGP2 had significantly enhanced α-glucosidase activity. The product after gastric digestion had the strongest inhibitory effect, its ability to inhibit α-glucosidase activity was increased by about 45.87%compared with undigested PGP2 at the same concentration. The digestion product of pancreatic juice also inhibited the activity of α-glucosidase by about 29.81% compared with undigested PGP2.This might be due to the digestion of pepsin and pancreatin, the sugar chain on the glycoprotein was released. According to research, some polysaccharides have a good inhibitory effect on α-glucosidase.According to the results in Fig. 16, we found that the digestion process weakened the inhibitory effect of PGP2 on α-amylase activity. Pepsin specifically acts on hydrophobic amino acids such as Phe, Trp, Tyr,and the sites of pancreatin are Argand Lys. The inhibitory activity of α-amylase might be related to these amino acid components. The inhibitory effect of PGP2 digested by pancreatic juice on α-glucosidase and α-amylase was weaker than that of products digested by stomach,the reason might be that the released sugar chains were partially hydrolyzed during pancreatic digestion. In summary, PGP2 can maintain strong anti-diabetic activity after digestion.

Fig. 15 Inhibition of PGP2 on α-glucosidase activity. The different letters means there was a significant difference between the two sets of data (P < 0.05).
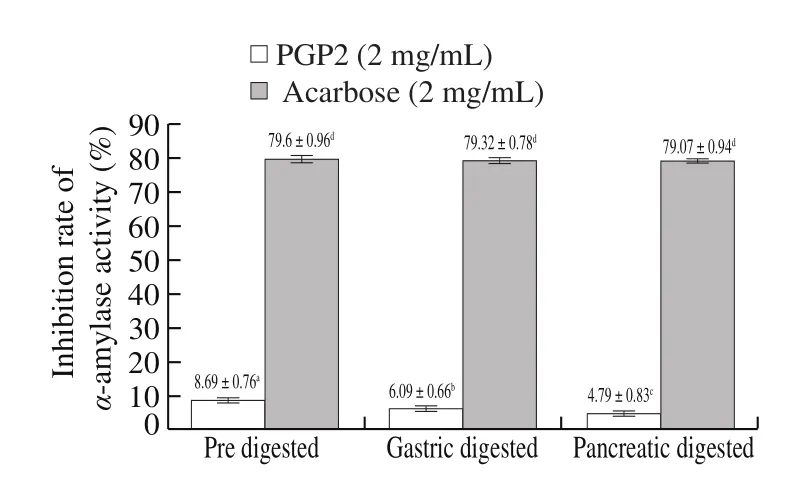
Fig. 16 Inhibition of PGP2 on α-amylase activity. The different letters means there was a significant difference between the two sets of data (P < 0.05).
4. Conclusion
In summary, 3 PGPs were separated and purified by a DEAE-Sepharose column and a Sephadex G-100 column. The physicochemical characteristics of 3 PGPs were evaluated and compared. The proteins and carbohydrates in these 3 fractions were connected by O-bonds. PGP2 had the highest protein content,while PGP1 had the highest polysaccharide content. The surface morphology of PGP1 and PGP2 was block-shaped and rod-shaped,while the structural morphology of PGP3 was mainly small spherical.After screening in vitro hypoglycemic experiments, we found that PGP2 in the 3 samples had the best hypoglycemic activity. It might be related to its higher glucose and arabinose content and lower molecular weight. The composition of monosaccharides might be the most important factor affecting its hypoglycemic activity. The reason for this speculation was that the inhibitory activity of PGP2 on α-glucosidase was significantly increased after digestion, while the inhibitory effect on α-amylase was slightly weakened. PGP2 can not only inhibit the uptake of glucose in small intestine, but also inhibit the activities of maltase and sucrase. Simulated gastrointestinal digestion test showed that PGP2 maintained strong hypoglycemic activity after digestion. Based on the study, PGP2 had the potential to be developed into a functional food or natural medicine to control blood sugar. This provides strong support for further research and application of PGP2 in the future.
Declaration of Competing Interest
The authors declare no competing financial interest.
Acknowledgments
The authors would like to show deepest gratitude tothe Earmarked Fund for Jiangsu Agricultural Industry Technology System(JATS[2020]413) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX19_0682)forfinancial assistance.

- 食品科学与人类健康(英文)的其它文章
- Moringa oleifera Lam. leaf extract mitigates carbon tetrachloride-mediated hepatic inflammation and apoptosis via targeting oxidative stress and toll-like receptor 4/nuclear factor kappa B pathway in mice
- Potential of peptides and phytochemicals in attenuating different phases of islet amyloid polypeptide fibrillation for type 2 diabetes management
- Zein as a structural protein in gluten-free systems: an overview
- Spectrum-effect relationship of immunologic activity of Ganoderma lucidum by UPLC-MS/MS and component knock-out method
- A red pomegranate fruit extract-based formula ameliorates anxiety/depression-like behaviors via enhancing serotonin (5-HT) synthesis in C57BL/6 male mice
- Effects of filleting methods on composition, gelling properties and aroma pro file of grass carp surimi