
生物素-硫胺素反应性基底节病的临床特点(附2例报告)
2021-05-19 08:38王明光张桐邓星强刘晓鸣
临床神经病学杂志 2021年2期
王明光,张桐,邓星强,刘晓鸣
生物素-硫胺素反应性基底节病(BTRBGD)自1998年报道第1例[1]至今,越来越多的病例被报道,其主要临床特征是肌张力障碍和对称性基底节病变,发病率不高,至今全球仅有百余例的病例报道,国内报道更是少见[2]。本文通过总结分析2例BTRBGD患儿的临床资料,以期对本病的诊断提供一定的借鉴作用。
1 临床资料
1.1 例1 患儿,女,10岁,因“阵发性抽搐10个月,呕吐6 d”于2017年3月30日入院。患儿生后6月龄时因反应差、喂养困难住院治疗,之后患儿出现运动发育停滞、肌张力增高表现,诊断“脑性瘫痪”行康复治疗。6岁10月龄时出现阵发性抽搐,表现为意识丧失、双眼凝视、牙关紧闭、口周发绀、四肢强直抖动,每次持续数十秒缓解,予口服卡马西平治疗后控制良好,抽搐未再发作。后自行停药,于2017年3月30日(患儿7岁8月龄)再次出现无热抽搐,入住本院神经内科。患儿出生史无异常,个人史无异常,家族史无异常。查体:神志清楚,反应好,头围正常,吐字不清,运动发育落后,不能独站及行走,四肢肌张力明显增高,腱反射活跃。头颅MRI示双侧基底节区对称性片状长T1、长T2信号,Flair及DWI序列呈稍高信号(图1)。拟诊线粒体脑病。检查线粒体基因未见异常,血常规、肝肾功能、心肌酶、电解质、血氨、乳酸未见异常。氨基酸有机酸代谢筛查未见异常。予口服丙戊酸治疗,未再抽搐,但肌张力增高及运动发育落后予康复治疗后无明显改善。
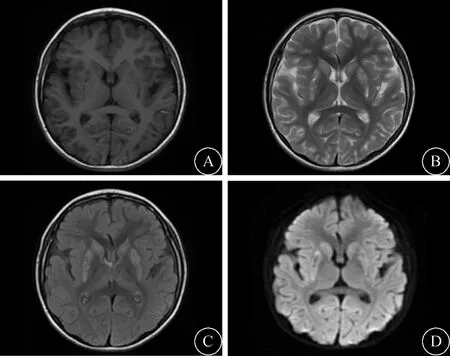
图1 例1患儿7岁8月龄时头颅MRI可见双侧基底节区对称性片状长T1长、T2信号,Flair及DWI序列呈稍高信号
1.2 例2 患儿,男,7岁,为例1的弟弟,因“阵发性头痛2个月”于2019年7月20日入院。患儿入院时为6岁5月龄,2个月前开始出现头痛,为阵发性两侧颞区痛,无发热、呕吐等伴随症状。患儿出生史无异常,个人史无异常,既往体健。查体:神志清楚,反应好,发育正常,四肢肌力及肌张力正常,腱反射正常引出,病理征阴性。头颅MRI检查示双侧基底节区见对称性片状长T1、长T2信号影,Flair及DWI序列呈稍高信号(图2)。查血常规、肝肾功能、心肌酶、电解质、血氨、乳酸未见异常。氨基酸有机酸代谢筛查未见异常。基因全外显子检测(2019-8-21)在SLC19A3基因发现3处致病突变,位置分别为chr2:228564172(c.259G>C p.G87R),chr2:228564017(c.414A>T p.R138S),chr2:228563844(c.587G>A p.S196N)。一代测序家系验证结果发现,染色体位置chr2:228564172,例1杂合突变,父亲杂合突变;染色体位置chr2:228564017,例1杂合突变,父亲杂合突变;染色体位置chr2:228563844,例1杂合突变,母亲杂合突变(图3)。父母均无临床表型,且非近亲婚配。

图2 例2患儿6岁5月龄时头颅MRI可见双侧基底节区对称性片状长T1、长T2信号,Flair及DWI序列呈稍高信号
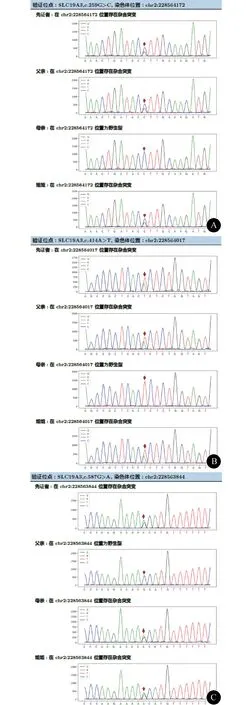
图3 BTRBGD患儿SLC19A3基因一代测序家系验证图。先证者:例2;姐姐:例1。A:染色体位置chr2:228564172,例1、例2杂合突变,父亲杂合突变;B:染色体位置chr2:228564017,例1、例2杂合突变,父亲杂合突变;C:染色体位置chr2:228563844,例1、例2杂合突变,母亲杂合突变
1.3 文献复习及诊治 本组患儿基因检测结果所示三个突变位点chr2:228564172(c.259G>C p.G87R),chr2:228564017(c.414A>T p.R138S),chr2:228563844(c.587G>A p.S196N)均未见文献报道(参考数据库:HGMD专业版数据库、Clinvar数据库)。三个突变位点分别来自父母,共同构成为复合杂合突变,符合BTRBGD隐性遗传模式。结合患儿的临床表型,病例1、病例2均诊断BTRBGD。予口服生物素3 mg/(kg·d),硫胺素逐渐加量至24 mg/(kg·d)。病例1患儿抽搐无发作,肌张力增高无明显改善,仍不能独站及行走。病例2患儿头痛缓解,未再发作。
2 讨 论
BTRBGD于1998年被阿拉伯人Ozand等[1]首次报道。本病多见于阿拉伯人近亲婚配孕育的子女,主要表现为肌张力障碍和对称性基底节区病变,对生物素治疗反应好,早期命名为生物素反应性基底节病,且仅强调生物素的治疗作用。随后Zeng等[3]研究发现SLC19A3基因是其致病基因。SLC19A3基因参与编码硫胺素转运体,且有报道部分患儿单纯应用生物素治疗效果差或病情复发出现急性代谢危象,联合硫胺素治疗后有效且无复发,更名为“生物素-硫胺素反应性基底节病”[4]。更有一些研究[5]提出单独应用硫胺素疗效与联合生物素、硫胺素疗效相当。
BTRBGD是一种常染色体隐性遗传病,位于染色体2q36.3上的SLC19A3基因为致病基因,该基因编码硫胺素转运体2,从而影响其在肠道、肝脏、脑等处的表达,所以补充外源性硫胺素治疗本病有效[6]。SLC19A3基因突变的表型差异很大,即便是同一家系相同基因型的患者,表型也可以有明显不同[4]。根据目前报道过的病例,表型主要有三种[7]。(1)经典型BTRBGD:儿童期起病,起病年龄5月龄~15岁,大多3~7岁起病,起病前发育正常,通常是由发热性疾病、创伤或手术等诱发,亚急性起病,可表现为意识障碍、构音障碍、吞咽困难、肌张力障碍、痫性发作、肢体活动障碍、甚至昏迷或死亡。痫性发作见于70%的病例,头颅MRI表现为基底节区的异常信号,可以累及皮质或皮质下白质,也可表现为慢性的基底节萎缩或坏死,对生物素、硫胺素治疗反应性较好[7]。(2)SLC19A3基因相关的Leigh样综合征和不典型婴儿痉挛:起病年龄在3月龄以内,Leigh样综合征的表现为呕吐、喂养困难、乳酸酸中毒及急性脑病的症状,头颅MRI可见对称性基底节病变;不典型婴儿痉挛的表现为痉挛发作,但EEG无高幅失律表现。这两种分型对生物素-硫胺素治疗反应性可能不好,预后较差[7]。(3)成人起病的SLC19A3基因相关的Wernicke’s样脑病:多成年起病,表现为癫痫持续状态、复视、眼球震颤、眼肌麻痹、共济失调等,头颅MRI可见两侧基底节区异常信号,可累及中脑导水管周围灰质,对大剂量的硫胺素治疗反应性好[7]。
本报道2例患儿为同胞姐弟,且基因一代测序验证结果突变位点相同,但表型明显不同,根据起病年龄和临床特征,结合以上表型分类,均为经典型BTRBGD。例1起病早,有喂养困难、运动发育落后、肌张力障碍、痫性发作等表现,结合头颅MRI对称性基底节区病灶,病程早期曾考虑为线粒体脑病(Leigh综合征),查线粒体基因结果正常。由于对本病认知不足,并未进一步完善全外显子检测,分别拟脑性瘫痪和癫痫进行康复治疗和抗癫痫治疗,但康复治疗效果差,肌张力障碍无好转。而痫性发作对抗癫痫药物治疗反应较好,口服丙戊酸或卡马西平均能有效控制痫性发作,这与Eichler等[8]研究结果一致。直至例2因反复头痛发作查头颅MRI发现类似的对称性基底节病变,且对BTRBGD疾病的认识逐渐提高,查全外显子基因检测结果SLC19A3基因突变,2例患儿均明确诊断为BTRBGD。予生物素、硫胺素治疗后例2头痛缓解。例1由于早期曾被诊断为Leigh综合征,未能早期明确诊断并及时给予生物素、硫胺素治疗,目前症状改善不明显,遗留了明显的肌张力障碍和轻度的认知功能障碍。此前Ortigoza-Escobar等[9]已报道本病早期易被误诊为Leigh综合征,且有报道[10]认为本病延误治疗可引起严重后遗症。例2以头痛为主要表现,且对生物素-硫胺素治疗反应好。以单纯头痛为主要表现的BTRBGD此前未见文献报道,本报道扩大了BTRBGD的症状谱,头痛可以为BTRBGD的首发表现。BTRBGD作为一种可治疗的代谢性疾病,早期诊断和治疗至关重要,早期应用大剂量的生物素和硫胺素治疗可以明显改善预后,Alfadhel等[7]推荐剂量为生物素5~10 mg/(kg·d),硫胺素10~40 mg/(kg·d)。
综上所述,临床工作中发现儿童不明原因的脑病伴有基底节对称性病变尤其是两侧壳核、尾状核病变,除了考虑线粒体脑病,还应考虑BTRBGD,尽快早期识别、完善基因检测明确诊断并给予治疗。
猜你喜欢
中国典型病例大全(2022年1期)2022-01-10
中国康复(2021年6期)2021-11-30
种子(2021年3期)2021-04-12
老年医学研究(2021年6期)2021-03-09
中外医疗(2018年12期)2018-09-03
家庭百事通·健康一点通(2017年8期)2017-08-18
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中西医结合心血管病电子杂志(2016年23期)2017-03-03
中国实用医药(2016年9期)2016-05-17
