
儿童线粒体脑肌病伴乳酸血症和卒中样发作误诊1 例临床特征并文献复习
2021-05-10 02:42李亚彬方辉张磊李岩
甘肃医药 2021年4期
李亚彬 方辉 张磊 李岩
阜阳市人民医院,安徽 阜阳 236004
众所周知,线粒体通过氧化磷酸化系统合成三磷酸腺苷(ATP)来提供机体的能量代谢,如果遗传基因突变引起线粒体结构和功能缺陷而导致ATP 合成障碍,就会造成机体能量代谢不足,尤其能量需求较高的脑组织、骨骼肌、心脏及肝肾等细胞能量代谢严重缺乏,引起线粒体脑肌病综合征,临床分为7 种类型[1]:线粒体脑肌病伴乳酸血症和卒中样发作(MELAS)、慢性进行性眼外肌瘫痪(CPEO)、KEARNS-SAYRE 综合征(KSS)、肌阵挛性癫痫伴肌肉破碎红纤维(MERFF)、肌神经胃肠型脑病(MNGIE)、LEBER 遗传性视神经病(LHON)及亚急性坏死性脑脊髓病(LEIGH),其中MELAS是线粒体脑肌病综合征中最常见的一种亚型[2],临床表现为复杂多样的多系统损害,缺乏特异性,易被误诊为脑炎、癫痫、脑梗塞、结节性硬化、脑瘫、矮小症等。现就我院1 例MELAS 综合征2 次误诊为病毒性脑炎的患儿报告分析如下。
1 资料与方法
1.1 一般资料 患儿女性,12 岁,因发热、呕吐伴精神差2 天于2020 年3 月24 日入住我院,体温最高38.7℃,发病前喝过凉饮料,发病后不能进食水,无抽搐发作及意识改变。G1P1 足月自然分娩,无产伤及窒息史。3 个月抬头,18 个月会走,2 岁前智力运动发育正常,以后智力、生长及语言发育逐渐落后于同龄儿童,但无惊厥史,现小学一年级,学习成绩很差。父亲肢体残疾,母亲智力障碍,非近亲结婚。入院后体格检查:体温38℃,营养发育差,身材矮小匀称,体重18kg,身高128cm,神志清楚,颈软,精神差,表情呆滞,不能行走,全身多毛、浓黑,以背部及双下肢显著,右眼外斜视,心肺正常,肝脾不大,四肢肌力肌张力正常,脑膜刺激征及锥体束征阴性。诊断“病毒性脑炎、营养不良、智力低下”给予阿昔洛韦、甘露醇、补液对症治疗8 天,体温正常、呕吐止、精神好转并能独自行走而出院。患儿间隔2 个月后无诱因下又出现发热、呕吐、头痛、精神差及不能行走,伴双手抖动、精神异常、小便失禁再次入院,又按脑炎治疗无好转,结合两次病史特点及辅助检查结果,考虑MELAS 综合征,给予对症、地塞米松等治疗13 天症状好转出院。
1.2 方法 对该患儿进行血液生化、神经肌电图、心电图、脑电图、头颅CT、头颅核磁平扫及血管成像检查、新生儿遗传代谢高相液相色谱-串联质谱检测以及基因突变检测和基因测序验证(从尿液中提取基因组DNA,PCR 扩增相关基因外显子及旁侧内含子区域,然后进行Sanger 测序,由福瑞医学检验实验室fulgent 基因完成)。
2 结果
2.1 实验室检查 脑脊液乳酸5.3mmol/L(正常范围0.6~2.4mmol/L),脑脊液细胞数偏高 19×106/L,糖及氯化物等正常,脑脊液培养阴性。血乳酸7.6mmol/L(正常范围 0.60~2.40),血常规、血糖、肝肾功能、心肌酶、电解质、免疫球蛋白、单纯疱疹病毒抗体、降钙素原、血沉、抗O 类风湿因子、抗核抗体、ANCA 抗体、血培养、PPD 试验、随机血生长激素均正常。两次住院检查流感病毒抗体IgM(+),促甲状腺素TSH0.219uIU/mL(参考范围 0.640-6.270)、游离 T3 2.39pg/mL(参考范围3.30~4.80)、游离 T4 1.57ng/dL(参考范围 0.86~1.40)。遗传代谢高相液相色谱-串联质谱法检测血精氨酸、亮氨酸、蛋氨酸、缬氨酸等多个氨基酸及游离肉碱、甲基丙二酰基肉碱增高。
2.2 脑电图检查 正常背景波消失伴一侧半球慢波活动。
2.3 神经肌电图 诱发电位提示所查肌肉未见明显神经源性及肌源性受累肌电图改变。
2.4 胸片及心电图 检查胸片阴性,心电图正常。
2.5 头部 CT、MRI 及 MRA 2020 年 3 月 25 日头部CT 灰白质对比正常,脑实质内散在多发斑片状低密度灶,第四脑室及枕大池扩大(图 1a)。2020 年 3 月 26 日头颅MRI 1.5T 平扫见两侧颞枕顶叶及右侧侧脑室体部旁斑片状、脑回样长T1 长T2,DWI 高信号,呈脑梗死改变。2020 年5 月24 日复查头颅MRI 1.5T 平扫见病灶侵犯左侧颞顶枕叶、左侧丘脑,右侧颞叶、右侧外囊区(图 1b、1c)。2020 年 5 月 6 日头颅 MRA 1.5T 血管成像相应区域未见明显异常(图1d)。
2.6 患儿及母亲基因检测结果 患儿DNA 线粒体m.3243A>G 基因位点存在突变(图2),杂质性变异比例为76.54%,纯质性变异比例为100%。患儿母亲DNA中线粒体基因m.3243A>G 未存在变异。
3 讨论
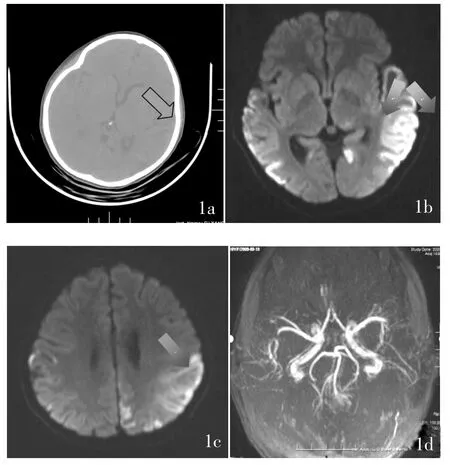
图1 患儿头颅CT 及头部核磁特征
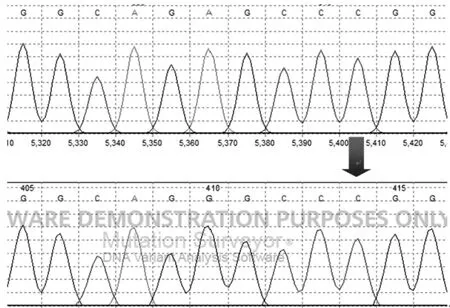
图2 尿液中线粒体基因mtDNA A3243G 突变位点
MELAS 综合征任何年龄均可发病,主要集中在2~40 岁,主要表现为青少年期卒中样发作的癫痫、头痛、呕吐、偏盲、痴呆、精神症状、听力下降、身材矮小及合并线粒体糖尿病[3]。Thorburn DR[4]报道 MELAS 综合征发病率约为1∶5000,我国儿童中的发病率尚不明确,仅见个案及小样本报道,但临床并非罕见,主要由于该病临床表现缺乏特异性,对此病缺乏足够的认识,基层医疗单位受条件限制不能进行肌肉活检及基因检测明确诊断,早期极易误诊为其他疾病。本例患儿系12 岁女童,急性发病,病程短,两次均以发热、头痛、呕吐、精神差为首发症状起病,伴行走困难,肢体抖动、精神异常及尿失禁,查体锥体束征及脑膜刺激征阴性,外周血炎性指标不高,脑脊液常规及生化仅细胞数轻度增高,脑电图正常背景波消失伴一侧半球慢波活动,存在脑功能损害,临床表现酷似病毒性脑炎及免疫性脑炎[5]。但间隔2 个月两次患同样的“脑炎”不符合常理,患儿无产伤、窒息史,2 岁前智力运动发育正常,以后智力、生长及语言发育逐渐落后于同龄儿童,身材矮小消瘦,多毛,眼外肌麻痹,呆容,结合母亲智力障碍,血及脑脊液乳酸均明显增高,头颅MRI 呈脑梗死改变,符合Kirino和Suzuki 提出的 MELAS 综合征诊断标准[6]:(1)28 岁前出现的脑卒中样发作。(2)具有以癫发作或痴呆为特征的脑病表现或两种表现同时存在。(3)存在高乳酸血症或破碎红纤维或者两种征象同时存在。(4)早期发育正常。(5)反复头痛。(6)反复呕吐。该患儿两次住院检测流感病毒抗体阳性,流感可引起发热、进食减少,能量代谢增高致使ATP 相对产生不足,加重线粒体的负担,氧化应激产生过多的氧自由基,导致线粒体衰竭而死亡,但流感病毒感染是否直接诱发MELAS 急性发作,临床鲜有报道;患儿甲状腺功能TSH 及T3 降低,可能是由于病灶累及丘脑,引起下丘脑促甲状腺激素释放激素分泌减少,进而造成脑垂体分泌促甲状腺素减少,甲状腺分泌游离T3 减少,这可能是生长发育迟缓、身材矮小原因之一;遗传代谢高相液相色谱-串联质谱法检测血精氨酸、亮氨酸、蛋氨酸、缬氨酸等多个氨基酸及游离肉碱、甲基丙二酰基肉碱增高,说明MELAS 综合征线粒体能量代谢障碍的同时可以合并多种氨基酸及有机酸的代谢异常。张金花等[7]报道6 例儿童MELAS容易误诊病毒性脑炎的原因[7]:(1)相同临床表现(发热、头痛、呕吐、抽搐);(2)相似的脑电图(背景慢化);(3)相同病变部位(海马、颞叶内侧面、岛叶、额叶眶面)。但病毒性脑炎好发于边缘系统,颞叶、额叶、岛叶皮层、扣带回多见,而枕叶、顶叶少见,多累及一个脑叶,常呈对称性[8];也可累及到脑实质深部,一般止于壳核外侧缘,较少向前或向后进展,可见轻至中度占位效应,而MELAS 综合征病灶不符合大血管分布,可资鉴别[9]。该患儿头颅核磁平扫显示脑部病灶多叶受累,分布在颞、顶、枕叶及丘脑、外囊区、侧脑室体部旁等多发对称性片状异常信号,T1 呈低信号,T2 呈高信号,额叶、岛叶皮层未受累,深部白质正常,不符合病毒性脑炎影像学表现;结合头颅MRA 血管成像相应区域无异常病变,不按解剖血管支配区分布,影像学改变符合MELAS 表现,而脑梗死病变主要累及血管分布区内的灰质和白质,多为单侧不对称性,也是与之相鉴别的重要依据。确诊还需要肌肉病理检查或遗传基因检测,由于家长及患儿原因,未能行肌肉活检,但基因检测患儿DNA 线粒体m.3243A>G 基因位点存在突变,杂质性变异比例为76.54%,纯质性变异比例为100%,符合文献报道[10],80%的MELAS 综合征是由mtDNA上第3243 位编码tRNALeu(UUR)的碱基发生腺嘌呤(A)到鸟嘌呤(G)的点突变(A3243G)引起,另外还发现[1]其他基因位点如 3271、3252、3231、11084 A→G 突变也可导致MELAS 综合征。韩萧迪等[11]报告29 例MELAS 综合征基因变异位点有26 例为m.3243A>G,占 89%,其余 3 例基因突变点 m.3271T>C、m.3481G>A、m.3946G>A 各 1 例。MELAS 综合征有 3 种遗传方式[12]:母系遗传(为线粒体基因突变所致)、常染色体隐性遗传和X 连锁遗传(为核基因突变遗传),本例报道患儿母亲DNA 中线粒体基因m.3243A>G 未存在变异,但母亲智力障碍,可能为X 连锁遗传。冯杰等[13]报道4例MELAS 综合征患儿基因检测证实均为线粒体DNA A3243G 突变引起,而且都携带母系突变基因,临床表型异质性多因DNA 突变的“异质性”和“阈效应”所致。总之,MELAS 综合征误诊的主要原因是仅停留在表象诊断,如脑炎、智力障碍、癫痫、脑性瘫痪、脑梗塞、结节性硬化、矮小症、营养不良、精神异常、共济失调等,没有详细询问围生期病史及母系家族史,运动障碍随饥饿或活动后加重,神经系统症状呈进行性加重,神经发育里程碑出现倒退,体检没有注意特殊外貌,如眼肌麻痹及多毛征等,忽视了头颅核磁多灶性脑损害、脑梗塞表现不符合大血管分布的特点,血和脑脊液乳酸增高未引起高度重视。随着广大医务工作者对此病的认识逐步深入,肌肉活检及基因检测逐步得到普及,就能够做到早发现、早诊断、早治疗,减少误诊,改善患儿整体预后。
猜你喜欢
临床肺科杂志(2022年3期)2022-11-26
中国现代医生(2022年19期)2022-11-04
中华实用诊断与治疗杂志(2022年1期)2022-08-31
现代临床医学(2022年1期)2022-02-12
中国卒中杂志(2021年7期)2021-11-29
健康大视野(2020年7期)2020-04-26
家庭科学·新健康(2019年1期)2019-03-06
中国体育科技(2018年6期)2018-12-13
东坡赤壁诗词(2018年3期)2018-07-16
扬子江(2016年1期)2016-05-19
