
一个甲型血友病家系新致病基因突变鉴定及分析
2021-04-29 09:45潘焯仪吴欣新张妙莲冼诗瑶吴海涛
福建医科大学学报 2021年1期
赵 强,潘焯仪,吴欣新,张妙莲,冼诗瑶,吴海涛
甲型血友病是一种常见的X染色体连锁隐性遗传病[1],在新出生男婴中的发生率约为1/5 000[2],其临床表现的严重程度具有较大差异性,主要是由血浆中凝血因子Ⅷ的功能活性所决定[3]。凝血因子Ⅷ是一种血浆糖蛋白,相对分子质量为267 kD,是凝血级联反应中一种重要辅助因子[4]。F8基因是凝血因子Ⅷ的编码基因,共包含26个外显子和25个内含子[5]。F8基因突变可造成凝血因子Ⅷ的数量或活性降低,从而影响凝血的级联反应,造成凝血功能障碍。目前,甲型血友病仍无有效的根治方案,只能通过定期补充凝血因子Ⅷ缓解病情的发展[6],通过孕前、胚胎前及产前诊断进行干预,预防患儿出生,可大大降低家庭及社会压力。因此,鉴定先证者致病基因突变就成为最关键的一步。
F8基因突变种类多样,已报道的突变包括错义突变、小插入缺失、倒位及拷贝数突变[7],还有部分患者无法找到致病基因突变,仅存在mRNA表达水平下降[8-9]。查询Human Gene Mutation Database(HGMD)可知,截至2019年1月,已报道的F8基因致病基因突变有3 000种以上,随着致病基因突变数据库的不断扩大,新突变报道的频率越来越低。本研究通过对一个甲型血友病家系进行F8基因突变的筛查,检测到一个未报道过的致病基因突变,并分析相关分子机制,确定其为该家系致病基因突变,并对2名该家系女性突变携带者进行产前诊断。
1 对象与方法
1.1对象 患者,女,29岁,孕14+3周,2017年 7月22日于笔者医院进行产前遗传咨询。孕妇的2个舅舅曾确诊为甲型血友病,有明确出血病史,实验室检测活化部分凝血活酶时间(activated partial thromboplastin time,APTT)延长,免疫检查为阴性。凝血因子活性检测,因子Ⅸ(factor Ⅸ,FⅨ)活性正常,因子Ⅷ(factor Ⅷ,FⅧ)活性明显降低,但家系所有成员并未进行基因诊断,家系成员关系见图1。本研究经笔者医院医学伦理会审核批准,患者知情同意。
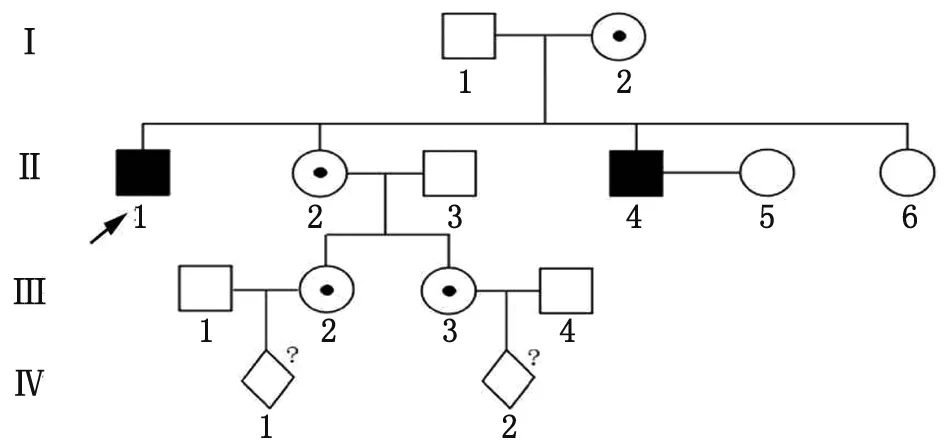
图1 甲型血友病家系图
1.2方法
1.2.1凝血功能检测 对该家系中所有成员采集外周血,进行凝血功能检测,检测指标包括凝血酶原时间(prothrombin time,PT)、凝血酶原时间国际标比率(international normalized ratio,INR)、APTT、纤维蛋白原(fibrinogen,Fib)、凝血酶时间(thrombin time,TT)、FⅧ活性、血浆D-二聚体(D-dimer,D-Di)。
1.2.2DNA提取及基因检测 对家系成员抽取外周血,采用DNA提取试剂盒(QIAamp DNA Mini Kit, 德国Qiagen公司)提取基因组DNA,对F8基因外显子及剪接位点区域使用特异性引物扩增,再行Sanger测序,测序数据通过与人类基因组参考序列(hg19)进行比对,鉴定出F8基因外显子区域及剪接位点的非同义突变及3个碱基内的插入缺失突变;采用多重连接探针扩增技术(multiplex ligation-dependent probe amplification, MLPA)(SALSA P178 F8 probemix, 荷兰MRC-Holland公司)对F8基因区域进行检测,检测有无50个以上碱基拷贝数的突变;使用重型血友病AF8基因22号内含子倒位基因检测试剂盒(深圳亚能生物技术有限公司)采用LD-PCR法[10-11]检测F8基因的22号内含子倒位。
1.2.3基因测序数据分析 首先检测出患者的所有基因变异,在HGMD(http://www.hgmd.cf.ac.uk/ac/index.php)、1000 Genomes、Hapmap(http://www.internationalgenome.org)、Pubmed(https://www.ncbi.nlm.nih.gov/pubmed/)及万方(http://www.wanfangdata.com.cn/index.html)等数据库进行检索,判断该突变是否已报道,并在家系其他成员中验证该突变,判断基因突变与家系成员临床表型是否存在共分离。然后,在50例无血缘关系且凝血功能正常的个体中检测该基因突变,确定其在本地人群的发生频率。最后对该基因突变位点进行生物功能学分析,通过Sift(http://provean.jcvi.org/index.php)和Ployphen(http://genetics.bwh.harvard.edu/pph2/index.shtml)软件预测突变功能,并通过SWISS-MODEL Homology Modelling对突变后蛋白三维结构预测及新发突变位置的氨基酸在不同物种的保守性分析,分析该突变对蛋白质功能的影响。
1.2.4产前诊断 家系中2例男性胎儿Ⅳ1及Ⅳ2 分别在孕16周和17周行羊膜腔穿刺术取羊水20 mL,采用微量样品基因组DNA提取试剂盒(北京天根生化科技有限公司),按说明书提取DNA。进而对2例胎儿的羊水DNA及其父母外周血DNA检测c.529T>C突变,判断胎儿是否携带致病性突变,并对父母、胎儿进行STR位点检测分析,确定羊水DNA有无母血污染及标本混淆的状况。
1.2.5新生儿凝血功能检测 胎儿出生后半年左右,进行血常规及相关凝血功能检测,并定期随访。
2 结 果
2.1家系成员凝血功能检测 家系成员凝血功能检测发现,Ⅱ1和Ⅱ4凝血功能显著异常,其中凝血因子Ⅷ活力降低,分别为4.7%和5.2%,其余成员凝血功能均正常(表1)。
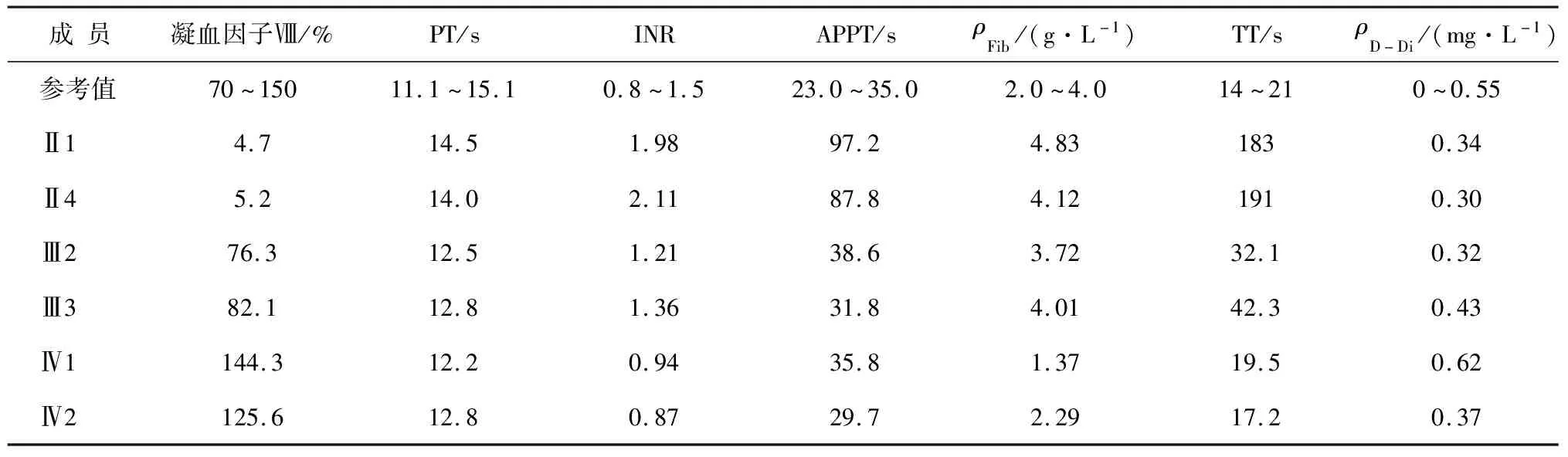
表1 家系成员的凝血功能检查
2.2对先证者进行F8基因检测 对Ⅱ1(男性患者)的F8基因外显子区域及剪接位点进行Sanger测序,检测出F8基因c.529T>C(p.Tyr177His)纯合突变(图2),考虑到该突变位于X染色体,男性仅有1条X染色体,所以应为该突变的半合子。MLPA结果显示,F8基因区域无插入缺失突变,也未检出22号内含子倒位。
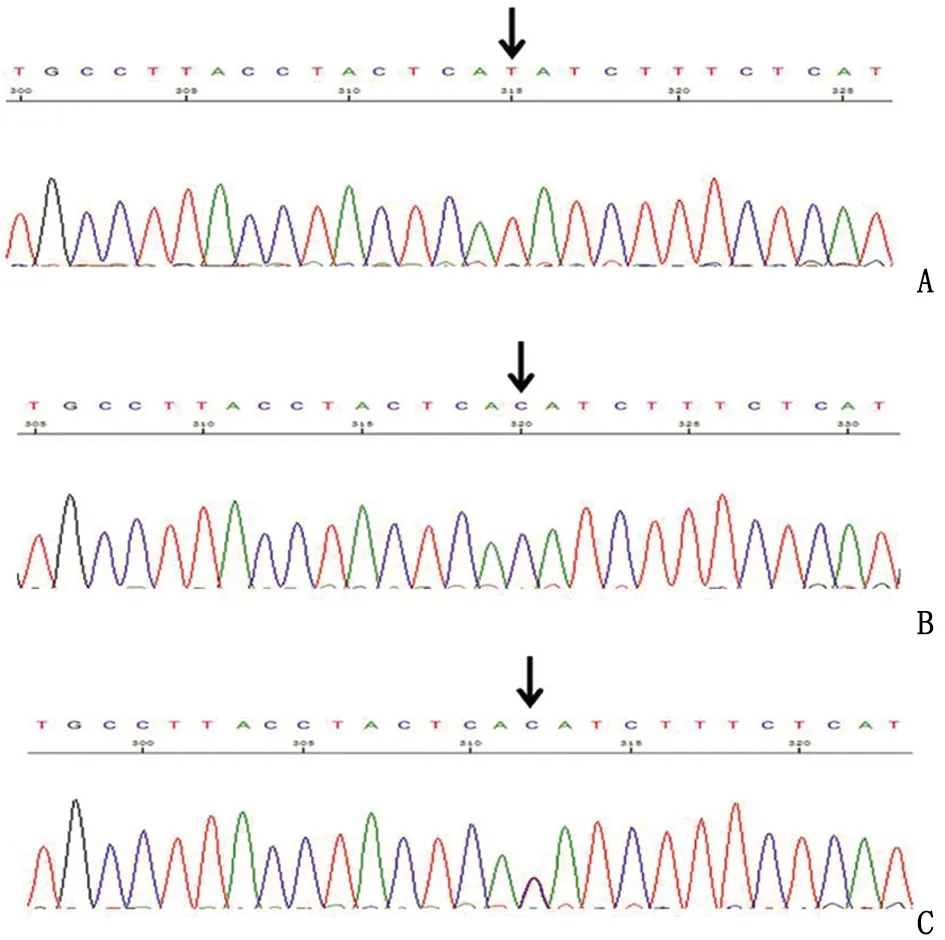
A:野生型位点测序图,家系成员Ⅳ1;B:纯合突变测序图,家系成员Ⅱ1;C:杂合突变测序图,家系成员Ⅲ2。
2.3文献及数据库检索 通过检索HGMD、1000 Genomes、Hapmap、Pubmed及万方等数据库,均未发现有报道该突变,证实该突变为新位点。
2.4家系基因与临床表型共分离分析 对家系中其他成员(Ⅰ2,Ⅱ2,Ⅱ4,Ⅱ6,Ⅲ2,Ⅲ3)通过Sanger测序对c.529T>C位点进行检测,2个男性患者(Ⅱ1和Ⅱ4)为c.529T>C半合子突变;Ⅰ2,Ⅱ2,Ⅲ2及Ⅲ3为c.529T>C突变杂合子携带者。该位点在该家系中符合基因突变与临床表型共分离。此外,在50例无血缘关系且凝血功能正常的女性个体中未检出该突变,确定其为罕见突变。
2.5生物信息学分析 通过Sift软件预测为“有害性突变”(得分值<10-3,阈值为0.05),Provean软件预测也是“有害性突变”(得分值-4.7,阈值为-2.5),Ployphen软件预测为“很可能致病性突变”,通过SWISS-MODEL Homology Modelling软件进行了突变蛋白三维结构预测(图3),发现该突变造成F8蛋白177位由酪氨酸变为组氨酸,由疏水性中性氨基酸变为亲水性碱性氨基酸,理化性质均发生了显著改变。同时,通过多物种序列比对(图4),F8蛋白177位氨基酸处于非常保守的状态。综合上述依据,判断该突变为可能致病性突变。

A:野生型F8蛋白结构;B:F8蛋白p.Tyr177His突变三维结构。红色圆圈内为177位氨基酸。

红色框内为177位氨基酸。
2.6产前诊断结果 家系成员Ⅲ2强烈要求进行产前诊断,通过与孕妇充分沟通,并签署知情同意书后,于孕17周抽羊水,通过Sanger测序,结果显示未携带有c.529T>C突变(图2A)。通过对父母胎儿DNA进行STR位点检测分析,确定羊水DNA无母血污染及标本混淆状况,并符合遗传规律,未发现羊水有母血污染干扰。胎儿Ⅳ1出生后7个月采静脉血,血常规及凝血功能检测结果均正常(表1)。
2018年3月,家系成员Ⅲ3也于孕16周要求行产前诊断,排查胎儿是否携带c.529T>C突变,结果显示也未携带该突变,胎儿Ⅳ2出生5个月采静脉血,血常规及凝血功能检测结果也正常(表1)。
3 讨 论
甲型血友病是一种常见的X染色体连锁隐性遗传病,是由F8基因缺陷导致凝血因子Ⅷ缺乏或功能缺陷所引起[12]。本家系Ⅲ2于孕14+3周时首次就诊,自诉具有甲型血友病家族史,主动寻求产前诊断,但该家族各成员均未进行过相关基因检测。本研究对该家系先症者Ⅱ1进行了F8基因常见遗传突变系统性筛查,检出一个新突变c.529T>C(p.Tyr177His)。为判断该突变的致病性,进一步检测该家系中各个成员该突变位点基因型,确定了c.529T>C在该家系中与个体的临床表型完全共分离,符合X染色体连锁隐性遗传规律。
本研究使用基因突变功能预测软件Sift和Polyphen对突变c.529T>C(p.Tyr177His)进行致病性分析,均提示该突变为有害性突变[13-14]。通过SWISS-MODEL Homology Modelling软件进行突变蛋白三维结构预测和F8蛋白多物种保守性分析,均表明该177位氨基酸具有重要功能性。再检索人类致病性突变数据库(HGMD)及已发表的中英文文献,均无该突变报道,确定其为新突变。有1例相似病例报道了c.530A>G(p.Tyr177Cys)突变,也导致F8蛋白的177位氨基酸改变,导致轻型甲型血友病[15],表明第177位氨基酸对维持蛋白功能活性具有重要影响。但此例c.530A>G(p.Tyr177Cys)突变是由酪氨酸突变为半胱氨酸,酪氨酸和半胱氨酸均为中性氨基酸,两者等电点分别为5.66和5.02。而本研究中检出的c.529T>C(p.Tyr177His)突变是由酪氨酸变为组氨酸,组氨酸属于碱性氨基酸,等电点为7.59。理论上可以推测该突变造成的理化性质变化比已报道的c.530A>G位点更加显著,造成的蛋白质功能改变同样也更显著。而实际上本研究中2个男性患者为c.529T>C(p.Tyr177His)半合子,凝血因子Ⅷ活力分别为4.7%和5.2%,临床表现符合中度血友病特征[16]。
本研究在该血友病家系中系统地对F8基因常见致病性突变进行检测,并通过可疑致病位点家系共分离检测和相应生物信息学方法分析预测,鉴定出F8基因c.529T>C(p.Tyr177His)为该甲型血友病家系疑似致病性基因突变。该新突变造成该家系2例男性患者表现为中度血友病特征,且为本研究首次报道。本研究对该家系中2个突变携带孕妇进行产前诊断,排除了2个男胎携带该致病性突变,2个男婴出生半年左右,凝血功能检测均正常,进一步证明了c.529T>C确实为该家系致病突变,本研究为F8基因致病突变谱贡献了一新突变,并明确了其致病性、患者凝血功能及临床表现。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
吉林林业科技(2018年6期)2018-11-21
中成药(2017年12期)2018-01-19
中国畜牧兽医文摘(2015年9期)2015-12-29
听力学及言语疾病杂志(2015年5期)2015-12-24
川北医学院学报(2015年5期)2015-12-05
西藏科技(2015年5期)2015-09-26
西南军医(2015年3期)2015-04-23
中国康复(2015年4期)2015-04-10
中国当代医药(2015年10期)2015-03-01
