
鼠伤寒沙门菌spvBC 基因编辑株的构建
2021-04-26 01:24:08左玲莉周丽婷吴兴旗吴超逸吴淑燕
生物技术通报 2021年2期
左玲莉 周丽婷 吴兴旗 吴超逸 吴淑燕
(1. 苏州大学医学部基础医学与生物科学学院,苏州 215123;2. 苏州高新区人民医院医学研究中心,苏州 215129)
沙门菌(Salmonella)是一类经粪口途径传播的革兰阴性菌,人和禽畜均可感染。2019 年加拿大六省沙门菌感染大暴发引起世界范围内的关注[1]。根据宿主感染后的临床表现,沙门菌可分为伤寒沙门菌(typhoidalSalmonella,TS)和非伤寒沙门菌(non-typhoidalSalmonella,NTS)两大类,前者主要包括伤寒和副伤寒沙门菌。除上述沙门菌以外的血清学归为NTS,在NTS 各血清型导致的全身感染中,鼠伤寒沙门菌(Salmonella typhimurium,S. typhimurium)是最常见的致病菌之一,约占65.2%[2]。免疫功能正常者感染鼠伤寒沙门菌多表现为胃肠炎型,免疫功能不全者则以败血症型和混合型多见[3]。我国感染性腹泻病因中鼠伤寒沙门菌感染占23.53%[4]。与此同时,鼠伤寒沙门菌血清型的不断变异和耐药菌株的相继出现为临床治疗增加了困难[5]。鼠伤寒沙门菌作为研究细菌致病机制与宿主相互作用的工具菌,已建立成熟的体内外模型[6]。因此,将其作为模式菌株,深入研究其致病机制并制定防控新策略具有重要的现实意义。
鼠伤寒沙门菌在与宿主的抗衡过程中,进化出一套精密的调控系统逃避宿主的免疫杀伤,其通过毒力岛2(Salmonellapathogenicity island 2,SPI-2)依赖的Ⅲ型分泌系统(type Ⅲ secretion system,T3SS)分泌的效应蛋白形成沙门菌包裹囊泡(Salmonella-containing vacuole,SCV),为自身复制和繁殖提供有利场所[7]。鼠伤寒沙门菌常含约8 kb的沙门菌质粒毒力基因(Salmonellaplasmid virulence gene,spv),该基因与细菌的毒力表型密切相关。spv基因由spvR、spvA、spvB、spvC和spvD五个开放阅读框组成[8]。其中spvB和spvC是研究最为广泛的两个毒力基因,与鼠伤寒沙门菌感染所致疾病的发生发展密切相关。spvB编码的效应蛋白SpvB 羧基末端结构域具有ADP 核糖基转移酶活性[9],能阻止肌动蛋白单体聚合,引起细胞骨架解聚[10-11]。本实验室前期研究证明在鼠伤寒沙门菌感染进程中spvB可抑制宿主细胞自噬,加重损伤[12]。spvC编码产物SpvC 具有磷酸苏氨酸裂解酶活性,可特异性地使丝裂原活化蛋白激酶(Mitogen-activated protein kinase,MAPK)信号传导通路不可逆的去磷酸化失活[13]。文献表明spvC可通过ERK 1/2 途径抑制肠道早期炎症反应,促进细菌全身播散[14]。本实验室研究发现spvC影响焦亡与其抑制肠道炎症促进细菌播散密切相关,但spvB与spvC的互作及其深入的致病机制尚未有文献报道。
λRed 同源重组系统是一种基于同源重组原理,对细菌染色体DNA 直接修饰的基因编辑系统,可在原核生物中实现快速的基因敲除。该系统需要pKD46、pKD4 和pCP20 三种质粒。pKD46 是λRed同源重组系统的协助质粒,L-阿拉伯糖诱导后可表达exo、bet和gam3 个基因编码的λ 噬菌体重组酶[15]。Exo 与Bet 引导线性片段与待敲除区域重组置换,Gam 抑制RecBCD 核酸外切酶的活性,使外源线性DNA 不至立即被降解[16]。pKD46 是温度敏感复制子,30℃培养可表达重组酶,37℃培养质粒则丢失。pKD4 为同源打靶片段提供卡那霉素(Kan)抗性基因的模板,两侧含翻转酶结合位点(Flipase recognition target,FRT),为重组转化体提供筛选标志。pCP20 编辑的翻转酶重组酶(Flipase recombination enzyme,FLP)可识别FRT 位点使之发生自身同源重组,从而消除一个FRT 位点及抗性基因[17]。pCP20 在42℃时诱导重组酶表达,同时质粒逐渐丢失。λRed 同源重组系统具有操作简便,同源重组效率高,且对菌株生长整体无影响等优点[18-19]。
pBAD/g Ⅲ表达系统由araBDA 启动子启动,带有g Ⅲ分泌信号和氨苄青霉素(Amp)抗性基因,L-阿拉伯糖能调控和诱导其表达,且表达产物羧基末端含有His 尾。这一标签不影响目的蛋白的结构和功能,有利于蛋白的鉴定和筛选[20]。一般条件下,重组质粒的稳定性受细菌代数影响,研究表明利用pBAD/g Ⅲ原核表达载体构建的重组菌连续传代30代后,质粒目的基因片段仍具有结构稳定性[21]。
鉴于此, 本研究利用λRed 重组系统和pBAD/g Ⅲ原核表达载体对spvBC进行基因编辑,构建鼠伤寒沙门菌质粒毒力基因spvBC敲除株和回补株,为深入探究鼠伤寒沙门菌致病机制及宿主的免疫应答提供可利用的工具。
1 材料与方法
1.1 材料
1.1.1 质粒和菌株 质粒pKD46、pKD4 和pCP20由普渡大学周道国教授惠赠。pBAD/g Ⅲ由江苏大学黄新祥教授惠赠。鼠伤寒沙门菌标准株野生型SL1344 由南京农业大学杨倩教授惠赠。
1.1.2 主要试剂和仪器 T4 DNA Ligase、XhoI 及EcoR I 限制性内切酶购自日本TaKaRa 公司;Easy Taq DNA Polymersae 购自中国全式金生物技术有限公司;Kod-Plus-Neo 高保真PCR 酶购自日本ToYoBo公司;鼠抗His 抗体购自优宁维生物技术有限公司;鼠二抗购自R&D Systems 公司;质粒小提试剂盒、通用型DNA 纯化回收试剂盒购自中国天根生化科技有限公司。PCR 扩增仪购自中国杭州朗基科学仪器有限公司;核酸电泳电泳系统购自中国上海天能科技有限公司;凝胶成像系统购自美国SYNGENE 公司;电转化仪购自美国Bio-Rad 公司。
1.2 方法
1.2.1 引物设计 利用GenBank 公布的鼠伤寒沙门菌spvB(ID:13909717) 及spvC(ID:13909727)基因序列,设计敲除引物H1P1和H2P2。5'端分别为50 ntspvB基因上游和spvC基因下游同源序列,3'端为pKD4kan抗性基因(两侧含FRT 位点)的上下游引物序列。此外,在spvBC基因的外侧设计一对鉴定引物P3和P4。该引物扩增产物中spvBC被kan替换后为1 747 bp,kan抗性基因消除后的敲除株中为356 bp。根据spvBC基因序列设计PCR 扩增引物spvBC-F(XhoI)和spvBC-R(EcoR I),并在引物5'端特异性酶切位点和保护碱基。引物序列见表1。
1.2.2 同源打靶片段的扩增 以pKD4 为模板,利用引物H1P1和H2P2PCR 扩增含spvBC同源臂及FRT 位点的kan抗性基因片段,PCR 反应体系:DNA 模板5 μL,引物H1P1和H2P2(10 μmol/L)各1.5 μL,Kod-Plus-Neo 高保真PCR 酶1 μL,10×PCR Buffer 5 μL,dNTPs 5 μL,Mg2+1 μL 去离子水30 μL。PCR 反应步骤:94℃ 5 min;94℃ 30 s,58℃ 30 s,68℃ 90 s,循环30 次;68℃ 7 min。利用DNA 纯化回收试剂盒纯化回收DNA 片段。

表1 实验所用引物序列
1.2.3 感受态的制备及Red 重组酶的诱导表达 鼠伤寒沙门菌SL1344 37℃、220 r/min 振荡培养过夜。按1∶50 转接菌液于100 mL LB 液体培养基中,37℃、220 r/min 振荡培养至OD600约0.6。将菌液冰浴20 min,在4℃条件下4 000 r/min 离心15 min,弃上清。加入冰浴的灭菌单蒸水10 mL 重悬,4 000 r/min 离心15 min,弃上清,重复洗涤3 次。再用冰浴的10%甘油10 mL 重悬,4 000 r/min 离心15 min,弃上清,洗涤1 次。最后加入冰浴的10%甘油500 μL 重悬,分装后于-80℃保存备用。
将感受态细胞冰上解冻,无菌电转杯冰上预冷。质粒pKD46 与感受态按1∶50 比例冰上混匀后进行电转。电转条件:电压2.5 KV,电容25 μF,电阻300 Ω,电转杯厚度2 mm。电转后立即加入预热到30℃的SOC 培养基混匀,30℃、220 r/min 振荡培养3 h。离心去上清,收集菌体,均匀涂布于含100 μg/mL Amp 的琼脂平板上,30℃培养过夜筛选阳性菌落。取新鲜单菌落接种于含3 mL 100 μg/mL Amp的LB 液体培养基,在终止培养前1 h 加入L-阿拉伯糖(终浓度30 mmol/L)诱导重组酶表达,再按照上述方式制备成含质粒pKD46 的感受态细胞。
1.2.4 同源打靶片段的电击转化及阳性克隆的筛选 取1 μL 纯化后的同源打靶片段电转入含质粒pKD46 的感受态细胞中,电转条件同1.2.3。电转后立即加入预热的SOC 培养基混匀,30℃、220 r/min振荡培养1 h 后37℃继续培养2 h。离心去上清,收集菌体,均匀涂布于含50 μg/mL Kan 的琼脂平板上,30℃培养过夜筛选阳性菌落。取单菌落利用引物P3、P4进行菌落PCR,鉴定完全重组的阳性菌落SL1344-ΔspvBC∷kan。
1.2.5 质粒pKD46 的消除 将阳性菌落涂布于Kan平板纯化3 次后于42℃培养过夜,将获得的单菌落分别涂布于含Amp 和Kan 的琼脂平板上,37℃过夜培养。在Kan 平板上生长而在Amp 平板上不生长的KanRAmpS菌落即为消除pKD46 的阳性菌落。
1.2.6 卡那霉素抗性基因的消除 将消除质粒pKD46 的阳性菌落按方法1.2.3 制成感受态细胞备用。将质粒pCP20 按1.2.3 方法电转入消除质粒pKD46 的感受态细胞中。产物涂布于Amp 平板,30℃培养过夜。将所得阳性克隆分别涂布于含Amp或Kan 的琼脂平板上,30℃过夜培养。在含Kan 的平板上不生长而在含Amp 的平板上生长的AmpRKanS菌落即为消除Kan 抗性的阳性菌落SL1344-ΔspvBC∷FRT。
1.2.7 质粒pCP20 的消除 将上述单菌落涂普通平板于42℃过夜培养以消除质粒pCP20。所得阳性菌落同时涂普通平板、Amp 平板和Kan 平板,30℃培养过夜,普通平板生长而抗性平板不生长得菌落为成功消除kan片段和质粒pCP20 的敲除株。连续传代3 次,利用引物P3、P4经PCR 鉴定spvBC基因敲除株SL1344-ΔspvBC。
1.2.8spvBC基因的扩增 以鼠伤寒沙门菌SL1344全基因组DNA 为模板,利用引物spvBC-F(XhoI)及spvBC-R(EcoR I)PCR 扩增含酶切位点的spvBC基因片段。PCR 反应体系同1.2.2。PCR 反应步骤:94℃ 5 min;94℃ 30 s,58℃ 30 s,68℃ 3 min,循环30 次;68℃ 7 min。利用DNA 纯化回收试剂盒纯化回收DNA 片段。
1.2.9 pBAD-spvBC重组质粒及回补株的构建 用限制性内切酶XhoI 和EcoR I 分别对上述PCR 扩增产物和质粒pBAD 双酶切,酶切产物经琼脂糖凝胶电泳鉴定,并切胶回收。将PCR 回收产物与载体质粒按体积比3∶1 混匀后经T4 DNA Ligase 在4℃连接过夜,最后将连接产物转化至DH5α 感受态细胞中,筛选阳性克隆。利用引物spvBC-F(XhoI)和spvBC-R(EcoR I)进行菌落PCR 鉴定,根据鉴定结果测序,与GeneBank 数据库中spvBC序列进行比对。将pBAD-spvBC重组质粒以电转化的方式导入鼠伤寒沙门菌spvBC基因敲除株内,操作步骤同1.2.3,涂布于Amp 琼脂平板筛选阳性克隆,连续传代3 次后所得单菌落即为鼠伤寒沙门菌spvBC基因回补株SL1344-c-spvBC。
1.2.10 Western blot 检测spvBC回补株SpvB 及SpvC蛋白的诱导表达 将回补株SL1344-c-spvBC接种到3 mL 含有Amp 的LB 液体培养基中,摇菌过夜培养,以SL1344-WT 和SL1344-ΔspvBC作为对照。取50 μL 菌液接种到5 mL 含Amp 的LB 液体培养基中继续培养2 h,再分别加入1.3 mmol/L 和13 mmol/L L-阿拉伯糖诱导继续培养3 h。将菌液4 000 rpm 离心5 min,弃上清,PBS 洗涤3 次。加入10 μL SDSPAGE 上样缓冲液,混合均匀,将样品置于100℃加热5 min,-20℃保存待用。以SpvB 抗体检测回补株SL1344-c-spvBC中蛋白SpvB 的表达,His 标签抗体检测回补株SpvC 蛋白(含His 标签)的表达情况。取30 μg 样品上样进行SDS-PAGE 电泳(浓缩胶60 V,约40 min;分离胶120 V,约40 min)。将蛋白湿转至PVDF 膜上(200 mA 恒流,60 min),置于摇床室温封闭1 h。用抗体稀释液稀释鼠抗SpvB、His抗体(1∶1 000 稀释),4℃孵育过夜。TBST 洗涤3次,每次10 min。抗体稀释液稀释山羊抗小鼠IgGHRP(1∶3 000 稀释),室温孵育1 h。TBST 洗涤3次,每次10 min。化学发光法显影,检测SpvB 及带His 标签的SpvC 蛋白表达水平。
2 结果
2.1 spvBC同源打靶片段的扩增
以pKD4 为模板,利用引物H1P1和H2P2PCR扩增spvBC同源打靶序列。该引物扩增含FRT 位点的kan片段为1 496 bp,加上两端50 bp 的spvBC同源臂,PCR 扩增产物应为1 596 bp。琼脂糖凝胶电泳显示扩增产物大小与理论值相符,表明成功扩增出含spvBC同源臂和kan抗性的DNA 片段(图1-A)。
2.2 spvBC同源打靶片段的转化与重组
将纯化后含同源臂的kanDNA 片段电转入含质粒pKD46 的感受态细胞中,涂Kan 平板,30℃培养,筛选出阳性菌落。利用引物P3、P4进行菌落PCR鉴定,基因spvBC被kan替换后为1 747 bp。琼脂糖凝胶电泳显示扩增产物大小与理论值相符,表明kan抗性成功替换spvBC基因,即获得完全重组菌SL1344-ΔspvBC∷kan(图1-B)。
2.3 kan抗性基因的消除及spvBC敲除株的鉴定
将完全重组的阳性菌落SL1344-ΔspvBC∷kan涂布于Kan 平板纯化3 次后于42℃培养过夜,筛选出KanRAmpS菌落即为消除pKD46 的阳性菌落。电转入质粒pCP20,筛选出AmpRKanS菌落即为消除Kan 抗性的阳性菌落SL1344-ΔspvBC∷FRT。再将其42℃培养16 h,筛选出AmpSKanS菌落即为消除质粒pCP20 的spvBC基因敲除株SL1344-ΔspvBC。连续3 次传代后,利用引物P3、P4进行PCR 鉴定,kan基因消除后敲除株中片段大小为356 bp。琼脂糖凝胶电泳显示扩增产物大小与理论值相符,表明成功构建spvBC基因敲除株SL1344-ΔspvBC(图1-B)。
2.4 spvBC基因的扩增
以鼠伤寒沙门菌野生株SL1344 全基因组DNA为模板,利用引物spvBC-F(XhoI)及spvBC-R(EcoR I)PCR 扩增含酶切位点的spvBC基因片段,相应引物扩增的spvA、spvB、spvC和spvD基因片段作为对照。PCR 扩增目的产物片段大小应为2 789 bp。琼脂糖凝胶电泳显示扩增产物大小与理论值相符,表明成功扩增出含酶切位点得spvBC基因片段(图1-C)。
2.5 pBAD-spvBC重组质粒的构建
spvBC基因片段和质粒pBAD 经XhoI 和EcoR I 双酶切后切胶回收,回收产物连接转化。随机挑选单菌落,利用引物spvBC-F(XhoI)和spvBC-R(EcoR I)进行菌落PCR,10 号为鉴定出的阳性菌落(图1-D)。将该菌落PCR 反应产物送测序,测序结果与GeneBank 数据库中spvBC序列进行比对,结果表明pBAD-spvBC重组质粒的构建成功。

图1 spvBC 基因编辑株的构建
2.6 鼠伤寒沙门菌质粒毒力基因spvBC回补株蛋白的诱导表达
以鼠伤寒沙门菌回补株SL1344-c-spvBC作为受试菌,敲除株SL1344-ΔspvBC做阴性对照,SL1344-c-spvB作阳性对照。经不同浓度L-阿拉伯糖诱导后Western blot 检测SpvB 蛋白(约68 kD)表达水平。结果表明,13 mmol/L L-阿拉伯糖诱导后,回补株SL1344-c-spvBC中SpvB 蛋白正常表达(图2-A)。以鼠伤寒沙门菌回补株SL1344-c-spvBC作为受试菌,野生株SL1344-WT 及敲除株SL1344-ΔspvBC做阴性对照。经不同浓度L-阿拉伯糖诱导后Western blot 检测回补株SpvC 蛋白上特有的His 标签(约30 kD)。结果表明,13 mmol/L L-阿拉伯糖可诱导回补株SL1344-c-spvBC中SpvC 蛋白正常表达(图2-B)。以上结果表明,鼠伤寒沙门菌回补株SL1344-c-spvBC中SpvB 及SpvC 蛋白L-阿拉伯糖适宜诱导浓度为13 mmol/L。
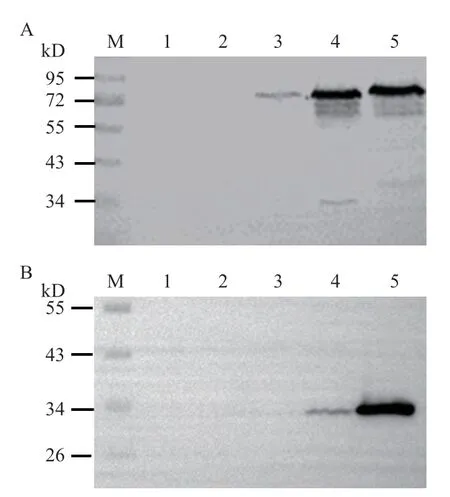
图2 spvBC 回补株蛋白的诱导表达
3 讨论
研究细菌基因功能的方法主要有基因敲除、反义RNA 干扰、转座子插入突变、基因过表达等,但因其余方法影响因素较多、插入位点不特异和筛选方法繁琐等缺点,基因敲除成为较可靠而常用的手段[22]。λRed 重组系统来自λ 噬菌体,克服了大肠杆菌自身核酸外切酶降解线性DNA 的缺陷,可实现几乎任何位点的基因修饰,且在子代中稳定遗传[23]。此外,λRed 重组系统不必依赖于酶切连接等传统的基因工程手段,达到近乎“无痕”的敲除效果。1998 年,Murphy 等[24]首次利用Red 重组系统在大肠杆菌中进行基因敲除,由此在原核生物中建立了新的基因编辑技术。随后广泛应用于大肠杆菌染色体基因的敲除[18],也有文献报道其用于沙门菌染色体基因的敲除[25],本实验室前期利用λRed 重组系统成功敲除鼠伤寒沙门菌质粒毒力基因spvC,但未用于长约2.8 kb 的大片段基因的敲除[26]。本研究利用λRed 重组系统敲除鼠伤寒沙门菌质粒上的大片段毒力基因spvBC,为研究大片段基因的功能提供工具,也为该系统在细菌质粒基因上的应用提供思路。
影响Red 同源重组效率的因素包括线性DNA的浓度、同源片段的长度、错配序列及内源性核酸酶等[27]。其中同源臂长度对重组效率至关重要。在大肠埃希菌中,同源臂从20 bp 增加至40 bp,同源重组效率呈指数级增长,而同源臂从40 bp 增加至1 000 bp,同源重组效率只增加10 倍。同源臂为40-50 bp 同源打靶片段即可进行高效率的重组[28]。有报道称600-1 000 bp 的同源臂,特别适用于鼠疫耶尔森菌大片段(2-47 kb)的基因敲除[29]。本研究发现,在本实验条件下,鼠伤寒沙门菌质粒毒力基因同源臂长度为50 bp 即可发生高效重组,既降低实验成本,简化实验操作又保证了敲除效率。本研究基于λRed 重组系统构建鼠伤寒沙门菌质粒毒力基因spvBC敲除株,为研究鼠伤寒沙门菌spvBC大片段基因的致病机制提供有力的工具。
原核生物中外源基因的存在犹如细胞内多拷贝质粒一样,对宿主是一种代谢负担。当外源基因片段较大且表达特异蛋白时,这种代谢压力就变得更加严重。研究人员将幽门螺旋杆菌HopE基因克隆到pBAD 载体上构建基因修饰菌,其编码外膜蛋白HopE 能稳定表达并保持较高拷贝数[30]。本研究利用pBAD 原核表达载体成功构建spvBC回补株,初步摸索了该系统的实验条件,优化回补株蛋白SpvB及SpvC L-阿拉伯糖诱导浓度。L-阿拉伯糖适宜诱导浓度为13 mmol/L,低浓度(1.3 mmol/L)L-阿拉伯糖诱导后SpvB 及SpvC 蛋白难以充分表达,而L-阿拉伯糖浓度较高(120 mmol/L)则抑制细菌生长[31]。
鼠伤寒沙门菌经粪口途径传播,在肠道内繁殖,粘附于肠黏膜上皮细胞,进而侵入固有层,释放毒素,导致其充血、水肿、点状出血等急性炎症反应[32]。本课题组新近研究表明,鼠伤寒沙门菌质粒毒力基因spvB通过核因子E2 相关因子2(Nuclear factor erythroid-derived 2-related factor 2,NRF2)调节细胞内的铁稳态从而影响细菌的胞内繁殖[33]。spvC可引起感染的免疫细胞逆向迁移进入血液,进而促进细菌的远处播散,导致败血症[34]。已知除鼠伤寒沙门菌外,其他致病沙门菌也大都含有spv基因,以上研究提示spvB及spvC作为沙门菌保守的毒力因子,在细菌的免疫逃逸和应对宿主固有免疫应答具有重要的作用。然而spvB与spvC的关系及其在感染进程中的互作仍需进一步探究。本实验构建鼠伤寒沙门菌质粒毒力基因spvBC敲除株和回补株,为后续深入研究spvB和spvC各自功能及其互作提供可利用的工具,也为其他原核生物的基因修饰提供科学依据和思路。
4 结论
本实验分别利用λRed 重组系统和pBAD 原核表达载体构建鼠伤寒沙门菌spvBC基因敲除株及回补株,并发现13 mmol/L 的L-阿拉伯糖为诱导鼠伤寒沙门菌spvBC回补株中SpvB 及SpvC 蛋白正常表达的适宜浓度。鼠伤寒沙门菌spvBC基因编辑株的成功构建为原核细胞大片段基因的编辑提供了参考策略,也为后续探究spvB和spvC在沙门菌致病中的功能提供可利用的工具菌株。
猜你喜欢
——紫 苏
河南农业(2024年1期)2024-01-19 01:56:54
华人时刊(2023年1期)2023-03-14 06:43:36
中国饲料(2022年5期)2022-04-26 13:42:36
汉字汉语研究(2021年2期)2021-08-30 08:58:46
中国动物传染病学报(2021年3期)2021-07-21 03:19:30
首都公共卫生(2018年5期)2018-11-21 07:02:02
河北书画研究(2016年3期)2016-04-28 08:55:35
食品安全导刊(2015年11期)2015-12-02 04:51:30
生物加工过程(2014年1期)2014-05-04 08:05:48
食品工业科技(2014年7期)2014-03-11 18:15:14
