
固相萃取净化-双填料反萃取-GC/MS法分析卷烟烟气中甲基丁香酚
2021-04-24 04:42廖惠云吴洋孙海平叶远青王晨辉曹毅张华朱怀远
烟草科技 2021年4期
廖惠云,吴洋,孙海平,叶远青,王晨辉,曹毅,张华,朱怀远
江苏中烟工业有限责任公司,南京市建邺区兴隆大街29号210019
随着公众对吸烟与健康问题的日益关注,卷烟烟气中释放的特定物质成分已成为关注的焦点[1-4]。在卷烟烟气中,存在着烯烃基苯类芳香物质,主要包含有丁香酚、甲基丁香酚、异丁香酚、甲基异丁香酚等,其分子结构中以苯环上有烯丙基或丙烯基为基础,链接有甲氧基或亚甲二氧基取代的官能团,该类化合物可能影响吸烟者的健康[5-6]。1964年,Rodgman等[7]收集多达0.5 kg的烟气粒相物,分离鉴定出甲基丁香酚;2001年,Hoffmann等[8]公布的69种卷烟烟气受关注成分清单中包含有甲基丁香酚;2002年,Rodgman等[9]对烟气中已报道的有害成分进行了总结,认定卷烟烟气中存在149种受关注成分,其中也包含甲基丁香酚。由此可见,烟气中的甲基丁香酚一直受到科技工作者的广泛关注。然而受限于烟气中甲基丁香酚释放量水平低,以及烟气复杂基质对目标物的分析干扰严重等因素的影响,准确测定甲基丁香酚的释放量是烟气分析领域一项极具挑战的工作[10]。
在国内外,甲基丁香酚在一些香料植物中常被鉴定和量化分析[11-14],但是缺乏对烟气中该成分的分析研究,仅有的报道是2000年Stephen等[15]采用固相萃取-GC/MS方法,对美国市场上销售的卷烟烟气中包含甲基丁香酚在内的8种烯烃基苯类芳香成分进行分析。但是基于对该方法的重复验证结果发现,甲基丁香酚在文献所采用的键合CN基团的固相萃取柱上保留性不强,达不到选择性洗脱的目的,从而无法准确定量目标物。为解决量化分析甲基丁香酚带来的技术挑战,本研究中甄选出合适的反相固相萃取柱,选择性洗脱目标物,再通过使用设计的双填料反萃取装置,从含大量水-有机溶剂溶液体系中反萃取目标物,进而采用GC/MS选择离子监测方法进行检测,定量分析出烟气中的甲基丁香酚,旨在为吸烟与健康研究提供科学依据。
1 材料与方法
1.1 材料、试剂和仪器
市售的28个国内外代表性品牌规格卷烟样品(其中样品1#~3#为混合型卷烟,样品4#~28#为烤烟型卷烟)。
甲醇、乙醇、正己烷和二氯甲烷(色谱纯,美国Sigma-Aldrich公司);甲基丁香酚(标样,≥98%)、3",4"-(亚甲基二氧)苯乙酮(内标,≥98%)(比利时Acros公司);超纯水(电阻率为18.2 MΩ·cm,自制);硫酸钠、硫酸镁、氯化钙和中性氧化铝(AR,使用前均经高温活化处理,国药集团化学试剂有限公司);4Å分子筛(使用前经高温活化处理,淄博齐创新材料科技有限公司)。
RM20H转盘式自动吸烟机(德国Borgwaldt KC公司);6890N/5975C气相色谱-质谱仪(配备7683自动进样器、增强型化学工作站,美国Agilent公司);Milli-Q超纯水系统(美国Millipore公司);AL204电子天平(感量0.000 1 g,瑞士Mettler Toledo公司);HY-8回旋式振荡器(常州国华电器有限公司);硅胶固相萃取柱(500 mg/6 mL)、Florisil固相萃取柱(1 g/6 mL)、氨基固相萃取柱(500 mg/6 mL)、氰丙基固相萃取柱(1 g/6 mL)、C18固相萃取柱(2 g/10 mL)、聚丙烯空柱管(带筛板/60 mL)(天津博纳艾杰尔科技有限公司)。
1.2 方法
1.2.1 标准溶液的配制
1.2.1.1 内标溶液
准确称取100 mg 3",4"-(亚甲基二氧)苯乙酮,用乙醇定容至100 mL,配制成浓度为1 mg/mL的内标储备液;准确移取100μL内标储备液,用乙醇定容至100 mL,配制成浓度为1μg/mL的内标溶液。
1.2.1.2 标准系列溶液
一级标准储备液:准确称取100 mg甲基丁香酚,用乙醇定容至100 mL,浓度为1 mg/mL;二级标准储备液:准确移取1 mL一级标准储备液,用乙醇定容至100 mL,浓度为10μg/mL。
标准系列溶液:分别准确移取二级标准储备液10、20、50、100、200和400μL,以及内标溶液各1 mL,用乙醇定容至10 mL容量瓶中。该系列标准溶液目标物浓度分别为10、20、50、100、200和400 ng/mL,其中内标浓度为100 ng/mL。
1.2.2 样品分析
1.2.2.1 样品萃取
按照GB/T 19609—2004[16]的要求收集20支卷烟的总粒相物。将捕集总粒相物的玻璃纤维滤片置于100 mL锥形瓶中(滤片应整片放入锥形瓶,铺平,不能剪碎或撕碎),准确加入40 mL甲醇,置于回旋式振荡器内,于160 r/min条件下振荡萃取40 min,静置。
1.2.2.2 萃取液净化
准确移取20 mL萃取液至浓缩瓶中,在70℃、常压下旋转蒸发至约1 mL,加入4 mL水,混合均匀形成乳化液,视为上样液。接着对上样液进行固相萃取(SPE)处理,具体步骤:用5~10 mL甲醇活化C18键合的SPE小柱,排干溶剂,再用10 mL水对SPE小柱进行平衡;然后移取全部上样液至SPE小柱上,待上样液流出SPE小柱后,分两次用20 mL 50%(体积分数)甲醇水溶液洗涤浓缩瓶,一并转移至SPE小柱内,弃去淋洗液;接着用10 mL 70%的甲醇水溶液洗脱,洗脱速度保持在1~2 mL/min,收集所有洗脱液,得净化液。
1.2.2.3 净化液反萃取
在一聚丙烯空柱管里,先加入6 g无水硫酸镁,再加入22 g无水硫酸钠,用吸耳球轻轻敲打,使其装填充实、均匀,然后对净化液中的目标物进行反萃取。具体步骤:加入10 mL二氯甲烷,使双填料物浸润;然后移入上述净化液,待净化液全部迁移进填料中时,用10 mL二氯甲烷洗涤盛装净化液的玻璃器皿,洗涤液全部转移至空柱管内,并用60 mL二氯甲烷分3次洗脱,使用双联球适当加压,收集缓慢流下来的溶液,得到反萃取液。
1.2.2.4 反萃取液浓缩及仪器分析
在反萃取液中加入100μL二级内标溶液,于40℃、常压条件下浓缩至约1 mL,进行GC/MS分析。分析条件:
色谱柱:DB-5MS毛细管柱(30 m×0.25 mm×0.25μm);进样口温度:250℃;进样量:1μL;分流方式:脉冲不分流;升温程序:60℃280℃(5 min);传输线温度:280℃;电离方式:EI;电离能量:70 eV;离子源温度:230℃;四极杆温度:150℃;检测方式:选择离子监测(SIM);目标离子:甲基丁香酚的特征离子为m/z 178、m/z 163和m/z 147(其中m/z 178为定量离子),内标的特征离子为m/z 164、m/z 149和m/z 121(其中m/z 149为定量离子),对每个离子的监测时间为100 ms;溶剂延迟时间:6 min。
2 结果与讨论
2.1 样品前处理条件优化
2.1.1 主流烟气捕集方式确定
卷烟烟气包括粒相和气相两部分,玻璃纤维滤片所捕集的仅是烟气的粒相部分。为了考察玻璃纤维滤片捕集主流烟气中甲基丁香酚的效率,利用其捕集主流烟气粒相物的同时,在滤片后串联两个盛有20 mL甲醇的吸收瓶,用于吸收主流烟气气相部分的甲基丁香酚。按照上述样品处理方法,抽吸20支卷烟,然后分别检测玻璃纤维滤片和吸收液中的甲基丁香酚。结果表明,玻璃纤维滤片捕集的甲基丁香酚的量为3.14 ng/支,而在两个吸收瓶的吸收液中均未检出甲基丁香酚。这说明主流烟气中的甲基丁香酚主要存在于粒相物中。因此,采用玻璃纤维滤片可以较为完全地收集卷烟主流烟气中的甲基丁香酚。
2.1.2 固相萃取柱填料选取
通过甲醇萃取烟气粒相物得到的萃取液,由于样品基质复杂,干扰严重,再加上甲基丁香酚的质量分数处于纳克级水平,如果对其直接进样分析或浓缩富集后进样分析,在色谱图中根本无法准确找到目标物。为此,在进样分析前必须先对萃取溶液进行净化处理,以排除基质干扰。在参考文献[17]的基础上,按照极性由大到小的顺序,依次选择硅胶固相萃取柱、Florisil固相萃取柱、氨基固相萃取柱、CN固相萃取柱和C18固相萃取柱为对象,考察目标物的保留情况,以便甄选出合适的填料,达到选择性洗脱甲基丁香酚的目的。图1a~图1e分别是以标样溶液为上样液,得到的淋洗液(上样后不加洗脱溶剂时,从柱子下筛板流下的液体)和洗脱液(加洗脱溶剂时,从柱子下筛板流下的液体)中目标物提取离子色谱图。
从图1中可知,极性强的硅胶、Florisil固相萃取小柱,对甲基丁香酚基本没有保留;极性适中的氨基小柱对目标物的保留也不强,极性适中的氰丙基小柱对目标物有部分保留作用,但淋洗液中目标物的质量分数还是远大于洗脱液中,这说明该类型小柱对目标物的保留也不佳;极性弱的反相C18小柱对目标物的保留最好,洗脱液中目标物的质量分数远大于淋洗液中,前者峰面积占整个目标物总峰面积的比例大于99%。事实上,从原理上进行分析,便能理解C18反相柱对目标物具有较好的保留作用:物理特性数据表明,甲基丁香酚的辛醇-水分配系数lgKOW为2.97。一般说来,按照lgKOW值分类大小原则,当lgKOW值>1时,物质呈现弱极性,lgKOW值越大,其极性越弱。根据相似相容原理,极性弱的C18反相柱理论上与目标物具有较强的相互作用力,进而对目标物有较好的保留作用。基于此,确定C18固相萃取小柱作为样品萃取液的净化柱。
2.1.3 固相萃取淋洗条件的优化
为了尽可能地排除烟气基质干扰,需对萃取液进行SPE净化处理。上样后,部分干扰物会与目标物同时保留在吸附剂上,这些干扰物中的一部分与吸附剂的作用力弱于目标物,而另一部分则强于目标物,所以在保证回收率要求的情况下,在增加淋洗液洗脱强度以最大程度洗掉保留性能弱的干扰物的同时,尽量降低洗脱液洗脱强度以最大限度使保留性能强的干扰物留在吸附剂上。为此,以标样溶液为上样液,分别用体积分数为5%、10%、20%、30%、40%、50%、60%、70%、80%、90%和100%的甲醇水溶液洗脱,各自收集洗脱液进行分析,建立淋洗曲线,以探讨淋洗液和洗脱液的合适浓度。目标物在C18柱子上的淋洗曲线见图2。可以看出,当甲醇浓度低于40%时,甲基丁香酚仍保留在柱子上,没有被洗脱下来;当浓度增加至50%时,有低于3%的目标物被洗脱下来,总的来看,洗脱能力还不是很强;当浓度增加至60%时,95.42%的目标物被洗脱下来;当浓度继续增加至70%时,98.68%的目标物被洗脱下来;随甲醇浓度继续增加,溶剂的洗脱能力更强,目标物基本全被洗脱下来。依据上述淋洗溶剂和洗脱溶剂极性强度选取的原则,在保证目标物回收率较好的情况下,选取50%甲醇水溶液为淋洗溶剂,选取70%甲醇水溶液为洗脱溶剂。在此基础上,以实际样品为分析对象,进一步优化并确认淋洗溶剂用量为20 mL、洗脱溶剂用量为10 mL。
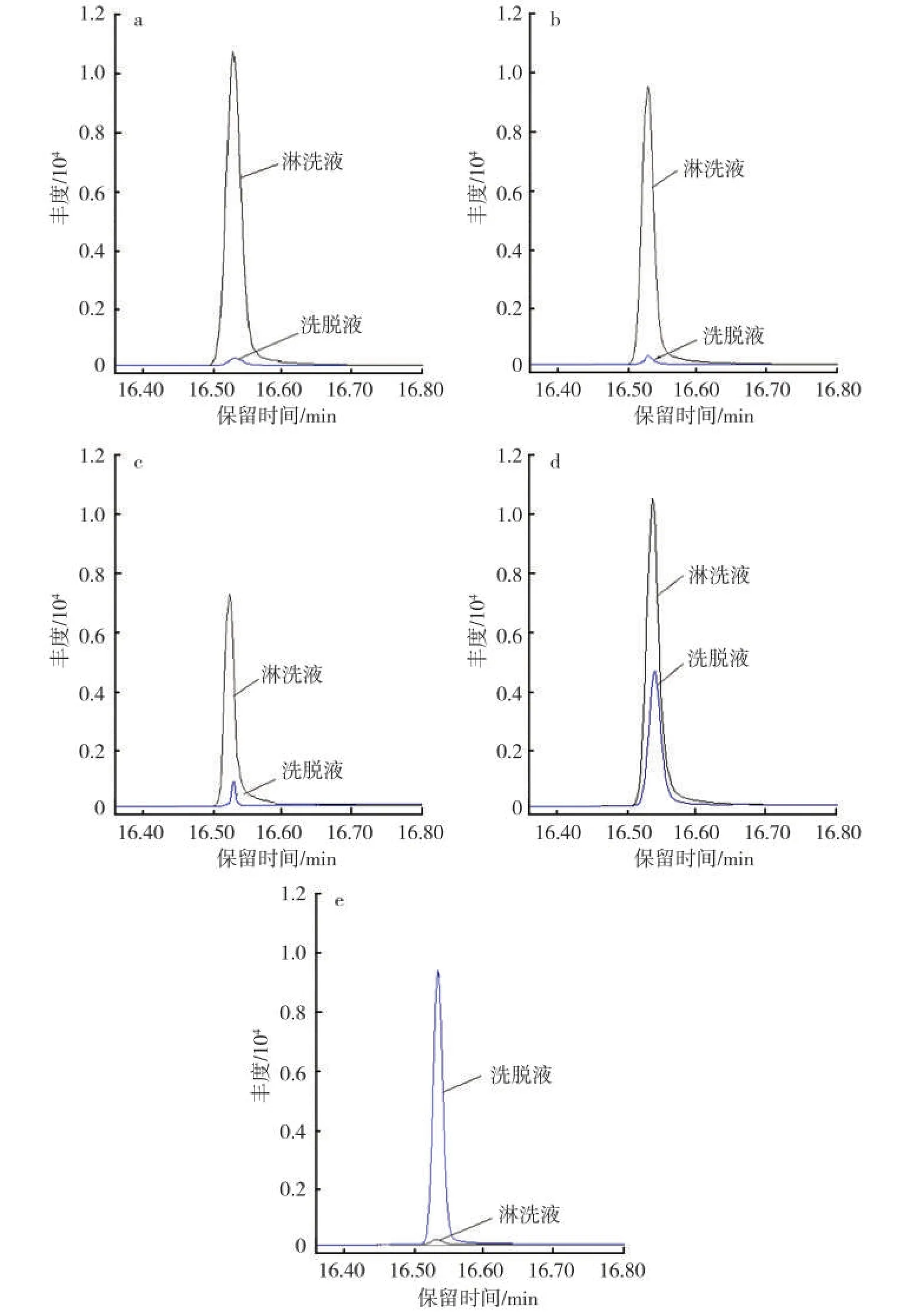
图1 不同SPE小柱条件下淋洗液和洗脱液中目标物的色谱图Fig.1 Quantitative ion chromatograms of methyleugenol in rinsing solution and elution solution by using SPE sorbents of silicone
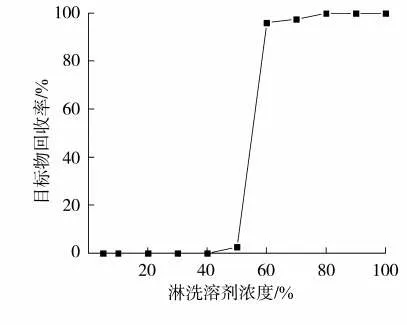
图2 目标物在C18柱上的淋洗曲线图Fig.2 Elution curve of methyleugenol by n-octadecane SPE
2.1.4 洗脱液除水反萃取的确定
经过C18SPE柱得到的净化液中含有一定量的水,理论上略大于洗脱溶剂中水的浓度(即大于30%)。在进行GC/MS分析前,必须进行溶剂转换,即采取相应的办法除去洗脱液中的水,选用合适的有机溶剂反萃取目标物,以避免水对目标物响应以及对色谱柱和质谱系统带来负面影响。为此,选取合适的溶剂转换办法就显得尤为重要。综合考虑到含水量较高以及目标物的质量分数极低,设计如图3所示的除水反萃取装置,并考察了硫酸钠、硫酸镁、氯化钙、中性氧化铝以及分子筛等对目标物出峰情况的影响。发现中性氧化铝以及分子筛对净化液的除水效果不佳,而氯化钙在甲醇环境下因会与其反应而不适宜除水。因此,确定选择硫酸钠和硫酸镁共同除水。依据硫酸钠吸水速度慢、吸附量大,以及硫酸镁吸水速度快、吸附量相对小的特点,将装置中的上填料确定为硫酸钠,下填料确定为硫酸镁。通过组合使用,充分发挥填料吸水特性,确保能够从含水量大的有机溶剂中反萃取出痕量的目标物。
使用该装置的反萃取过程为:先用二氯甲烷浸润双填料,使二氯甲烷充满整个双填料体系内;再添加洗脱液(约70%的甲醇水溶液),大部分水被上填料硫酸钠吸收,剩余的水被下填料硫酸镁快速吸收;同时,二氯甲烷因其密度比水大,夹带着甲醇慢慢从筛板下流出来;最后,使用大量的二氯甲烷进行淋洗,采用双联球略微加压,使目标物被溶剂淋洗下来,达到从含水量大的有机溶剂体系中反萃取痕量目标物的目的。

图3 洗脱液中除水反萃取装置示意图Fig.3 Schematic diagram of reverse extraction device for absorbing water in eluate
2.1.5 反萃取双填料使用量的优化
在优化洗脱液除水反萃取装置中,发现双填料的用量对反萃取效果影响明显:如果填料中硫酸钠用量过多、硫酸镁用量过少,使得含少量水的净化液不能快速被吸收而流出筛板,反萃取液中还含有微量的水分;如果填料中硫酸钠用量过少、硫酸镁用量过多,可使溶液体系内硫酸镁因吸水急剧放热,下填料会形成坚硬的块状固体,严重影响净化液的流出速度。为此,必须优化双填料的使用量。在前期反复实验的基础上,以10#卷烟样品为对象,按照表1所示的用量配比对填料用量进行优化。从结果(图4)可以发现,对于本样品前处理萃取液净化过程得到的净化液而言,硫酸钠-硫酸镁用量配比存在一个较好的平衡点,在m(硫酸钠):m(硫酸镁)=22:6时,既能保证洗脱液以一定流速从填料中流出,又能使得目标物的反萃取效果最优。因此,最终选择m(硫酸钠):m(硫酸镁)=22:6为双填料用量。
2.2 仪器分析条件选择
2.2.1 色谱进样方式的优化
基于目标物的痕量水平,为提高其响应情况,可使用脉冲进样模式,以提高实际进入色谱柱的样品量,减少其在进样口分解的机会,同时使样品更集中地进入色谱柱从而提高检测灵敏度[18]。为此,在保持其他色谱分析条件不变的情况下,研究了不同进样方式下目标物响应情况的变化,结果术分析烟气样品时,必须优选质谱特征离子。目标物甲基丁香酚的质谱图显示,其分子离子[M]+也是丰度最大的基峰离子为m/z 178,紧随其后的特 征 离 子 为m/z 163[M-CH3]+和m/z 147[M-OCH3]+,三者的丰度比为100:34:33;内标物3",4"-(亚甲基二氧)苯乙酮的质谱图显示其分子离子[M]+为m/z 164,紧随其后的特征离子为m/z 149[M-CH3]+,这也是丰度最大的基峰离子,再随其后的特征离子为m/z 121[M-CO-CH3]+,三者的丰度比为51:100:36。依据特征离子的选择应符合m/z大、相对丰度高、干扰离子少等原则,确定甲基丁香酚的定量离子为m/z 178,辅助定性离子为m/z 163和m/z 147;内标物的定量离子为m/z 149,辅助见图5。可以看出,4种进样方式下,目标物和内标的出峰情况均较好,峰形对称,基线完全分离。不分流(含脉冲)相比于分流(含脉冲)而言,前者的峰面积均大10倍多,与分流比大小基本相符合,同时信噪比也大3~4倍。进一步分析可知,在不分流条件下,相比于单纯的不分流,脉冲不分流条件下目标物和内标的峰面积和信噪比均比后者大。为此,考虑到目标物的整体出峰状况,优选脉冲不分流为色谱进样方式。
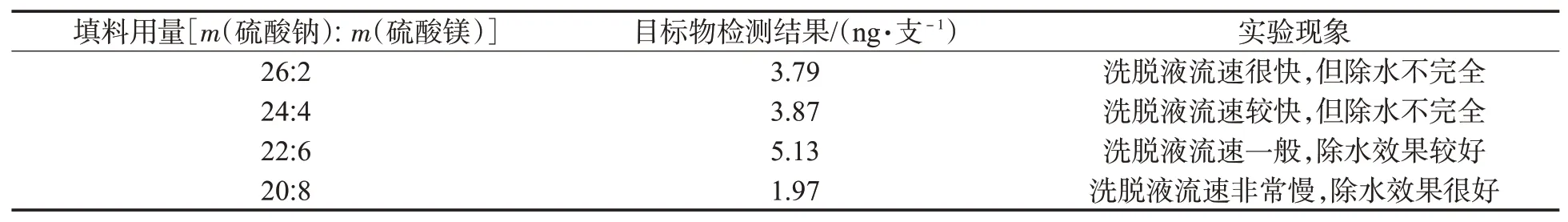
表1 不同填料用量对目标物反萃取的影响Tab.1 Effects of different usages of packing materials on reverse extraction of target compounds

图4 不同填料用量下目标物色谱图Fig.4 Quantitation ion chromatograms of methyleugenol at different ratios of packing materials
2.2.2 质谱特征离子的确定
基于卷烟烟气化学成分复杂、基质干扰严重及目标物释放量较低等因素,在使用SIM-GC/MS技定性离子为m/z 164和m/z 121。
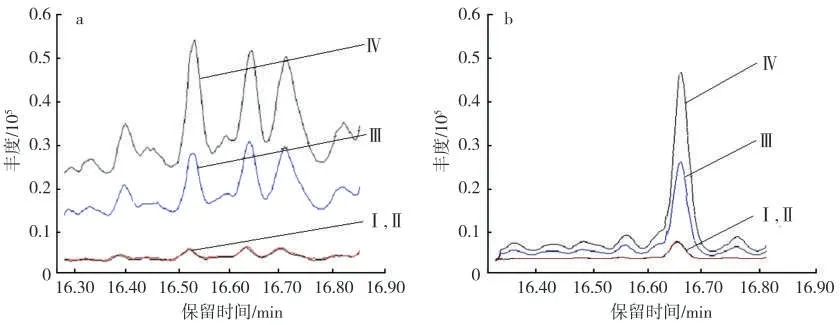
图5 不同进样方式下目标物(a)和内标(b)的提取离子色谱图Fig.5 Quantitation ion chromatograms of methyleugenol(a)and internal standard(b)in different injection modes
2.3 方法评价
2.3.1 工作曲线、检出限和定量限
将系列标准工作溶液进行GC/MS分析,求得线性回归方程及其相关参数。从表2可知,目标物在10~400 ng/mL浓度范围内(换算成烟支质量分数为1.00~40.0 ng/支)线性关系良好(回归方程的R2为0.999 5)。用最低浓度标样作为未知样品,重复进样10次,计算标准偏差。分别以3倍和10倍标准偏差计算方法的检出限(LOD)和定量限(LOQ)。LOD和LOQ分别为0.24和0.81 ng/支,远低于文献[15]的报道值(其检测限为5.1 ng/支),能够满足卷烟烟气中甲基丁香酚的检测要求。

表2 方法的工作曲线及线性范围、检出限和定量限Tab.2 Calibration curve,limit of detection,and limit of quantitation of the method
2.3.2 精密度
以14#卷烟样品为分析对象,分别进行日内和日间精密度实验,分别计算6次平行测定结果的相对标准偏差(RSD)。表3结果显示,方法的日内RSD<7%,日间RSD<9%。对于痕量物质分析而言,该方法精密度较好。
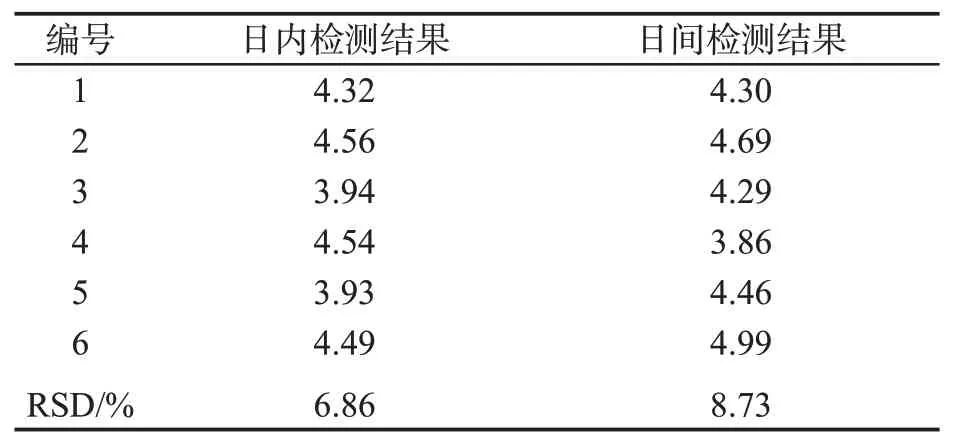
表3 方法的精密度Tab.3 Precision of the method (ng·支-1)
2.3.3 回收率
同样以14#卷烟样品为对象,进行低、中、高3个不同浓度水平的加标实验,然后按照上述前处理步骤处理,再进行GC/MS分析,计算其加标回收率。从表4可知,目标物的回收率在97.2%~114.5%之间。表明该方法的回收率较好。

表4 方法的回收率Tab.4 Spiked recoveries of the method
2.4 卷烟样品的测定
采用本方法分析28个卷烟样品的结果见表5。可知,在所测卷烟样品中,主流烟气甲基丁香酚的释放量范围为2.24~5.13 ng/支,其中混合型卷烟的释放量范围为2.46~3.52 ng/支,烤烟型卷烟的释放量范围为2.24~5.13 ng/支。从单位焦油释放量来看,混合型卷烟主流烟气中单位焦油甲基丁香酚释放量范围为0.45~0.82 ng/支,烤烟型卷烟的释放量范围为0.22~0.47 ng/支。综上,混合型卷烟主流烟气中甲基丁香酚的释放量与烤烟型卷烟相差不大,但对于单位焦油甲基丁香酚释放量而言,前者远高于后者。这与文献[19]中所述的丁香酚的释放规律一致,可能是因为该两个化合物均属于烯烃基苯类物质,主要来源于烟草中木质素燃烧产生的转移[7]。此外,值得说明的是,如若采用文献[15]的方法,其检测限为5.1 ng/支,远大于多数卷烟样品的实际释放量,根本无法检测出卷烟样品主流烟气中目标物的释放量,这也体现出本方法的优越性。

表5 28个不同品牌卷烟主流烟气中甲基丁香酚的释放量Tab.5 Releases of methyleugenol in mainstream cigarette smoke of 28 cigarette brands
3 结论
①建立了采用固相萃取净化-双填料反萃取-气相色谱/质谱法分析卷烟主流烟气中甲基丁香酚释放量的方法。②目标物在10~400 ng/mL浓度范围内线性良好,R2为0.999 5,日内和日间相对标准偏差分别为6.86%和8.73%,样品加标回收率在97.2 %~114.5 %之间,方法的检出限为0.24 ng/支,定量限为0.81 ng/支,本方法明显优于现有的国外文献报道结果。③部分样品普查结果表明,混合型卷烟烟气甲基丁香酚的释放量范围为2.46~3.52 ng/支,烤烟型卷烟烟气释放量范围为2.24~5.13 ng/支,其中前者单位焦油中的释放量远高于后者。本方法可以准确测定烟气中的甲基丁香酚释放量,可为有关吸烟与健康研究提供科学依据。
猜你喜欢
中国石油石化(2022年18期)2022-11-19
分子催化(2022年1期)2022-11-02
石油化工(2022年9期)2022-10-19
中国资源综合利用(2022年9期)2022-10-13
少年文艺(2022年8期)2022-07-08
绿色科技(2021年4期)2021-04-06
中小企业管理与科技·中旬刊(2018年8期)2018-11-12
中国经济周刊(2017年6期)2017-03-21
职工法律天地·上半月(2015年5期)2015-07-02
故事林(2010年23期)2010-05-14
