
鹿科动物基因组学研究进展
2021-04-22 07:37:18巴恒星胡鹏飞李春义
遗传 2021年4期
巴恒星,胡鹏飞,李春义
综 述
鹿科动物基因组学研究进展
巴恒星1,2,胡鹏飞1,2,李春义1,2
1. 长春科技学院,鹿茸科学与产品技术研究所,长春 130112 2. 吉林省鹿茸生物学重点实验室,长春 130112
鹿科动物是世界上最丰富的大型哺乳动物之一,在极北地区、热带地区和高海拔地区都有分布。中国占世界鹿科动物40%以上,是鹿科动物进化的主战场。鹿科动物除了具有反刍动物常见的独特表型特征外,更是进化出周期性再生新器官——鹿茸角。鹿科动物是研究生态学、行为学和进化生物学非常有价值的动物模型,特别是在研究哺乳动物器官再生方面具有重要科学价值。鹿基因组是系统阐述鹿的进化和演变,解析鹿科动物独特生物学性状的依据,对遗传资源保护和利用具有重要意义。目前,随着鹿科动物参考基因组的不断完善,在鹿基础科学研究上取得了诸多重要成果。本文详细综述了鹿科动物基因组学研究进展,主要包括鹿遗传变异数据、适应性进化分子基础、独特性状鹿茸角的起源进化关键基因和功能基因组学,以期为鹿科动物的深入研究奠定理论基础。
鹿科动物;参考基因组;遗传变异;适应性进化;鹿茸角;功能基因组学
鹿科(Cervidae)包括4个亚科,分别是鹿亚科(Cervinae)、獐亚科(Hydropotinae)、麂亚科(Muntiacinae)和空齿鹿亚科(Odocoileinae),共16属约52种,在反刍亚目里,种类上仅次于牛科(Bovidae),在极北地区、热带地区和高海拔地区都有分布。中国是鹿类动物最丰富的国家之一,占世界鹿科动物40%以上,是鹿科动物进化的主战场[1]。鹿科动物除了具有反刍动物常见的独特表型特征外,更是进化出周期性再生新器官——鹿茸角。科学家们普遍认为,鹿科动物是非常有价值的生态学、行为学和进化生物学的研究模型[1],特别是哺乳动物器官再生的研究模型[2],具有重要科学价值。
近年来高通量测序技术日新月异,推动了动物基因组学的飞速发展,基因组学研究方法也在不断地创新和改进,这些都为鹿科动物基因组学研究创造了前所未有的机遇,迎来鹿科学研究的新时代。目前,相关领域的研究人员已产生了大量鹿科动物的基因组学数据,在此基础上使解析鹿科动物的适应性进化分子基础、探究鹿茸角起源进化及鹿茸角再生发育相关通路和基因成为可能。不仅如此,基因组学研究还有利于加速家养鹿类动物品种培育和遗传改良进程。本文对近年来鹿科动物基因组学研究领域所取得的重要进展进行了系统地分析和总结,也对今后该领域研究方向进行了展望,以期为解析鹿科动物独特的生物学性状和遗传资源保护利用提供参考依据。
1 鹿科动物基因组资源
1.1 基因组参考序列
鹿科动物基因组测序起步较晚,2011年首次由美国贝勒医学院在NCBI GenBank上公开了白尾鹿()基因组,但未见文章发表。2014年也发布了西方狍()基因组Contig序列,仍未见文章报道。2017年,Li等[3]在和中国国家基因库(China National GeneBank, CNGB)上发表了驯鹿()基因组。通过构建9个不同长度插入片段测序文库,基于Illumina HiSeq 4000平台测序,组装2.64 Gb的驯鹿基因组草图,Contig和Scaffold N50大小分别为89.7 kb和0.94 Mb,注释了21,555个蛋白编码基因。随着基因组测序成本快速降低,最近3年,鹿科动物基因组序列发表数量迅速增加,已达16个鹿种(表1),涵盖了鹿科动物所有4个亚科,它们基因组组装大小在2.5~2.8 Gb之间,但这些鹿基因组仍处于草图水平,质量还有待进一步提升。为了更好地利用基因组数据解析鹿科动物的特殊表型性状,本文归纳了这16个鹿种的系统进化关系,并进一步总结了它们的染色体核型、形态学数据(图1)及自然地理分布(图2)。
目前,仅有5个鹿科动物的基因组组装到染色体水平,分别是塔里木马鹿()、赤鹿()、小麂()、赤麂()和梅花鹿(Nippon)。Ba等[4]利用由38,083个单核苷酸多态性位点(single nucleotide polymorphism, SNP)组成的高密度遗传连锁图谱[5]将塔里木马鹿基因组95.9%序列组装到34对染色体。其中,3号、8号和31号染色体仅由1个Scaffold组成。这34对染色体组装大小与遗传图谱评估大小相关系数高达0.987。由于构建大型野生动物遗传图谱十分困难,利用Hi-C技术组装基因组染色体也是目前最有力技术手段。Mudd等[6]利用Hi-C技术完成了小麂(2= 46)和赤麂(2=6♀/7♂)基因组的染色体组装。类似地,梅花鹿基因组也是通过Hi-C技术挂载到33对染色体上,但未见文章报道。
马鹿/赤鹿()是鹿科动物中最大的类群,由22个亚种组成,分布在欧洲南部和中部、北美洲、非洲北部和亚洲。在生态学、生物多样性和种间杂交等研究领域中,该类群研究最为广泛而备受关注。塔里木马鹿是中亚仅有的亚种,被认为是最原始亚种[7],有特殊的进化地位,其染色体水平基因组图谱将为马鹿/赤鹿类群,乃至整个鹿类动物的科学研究提供最有价值的参考基因组数据资源。
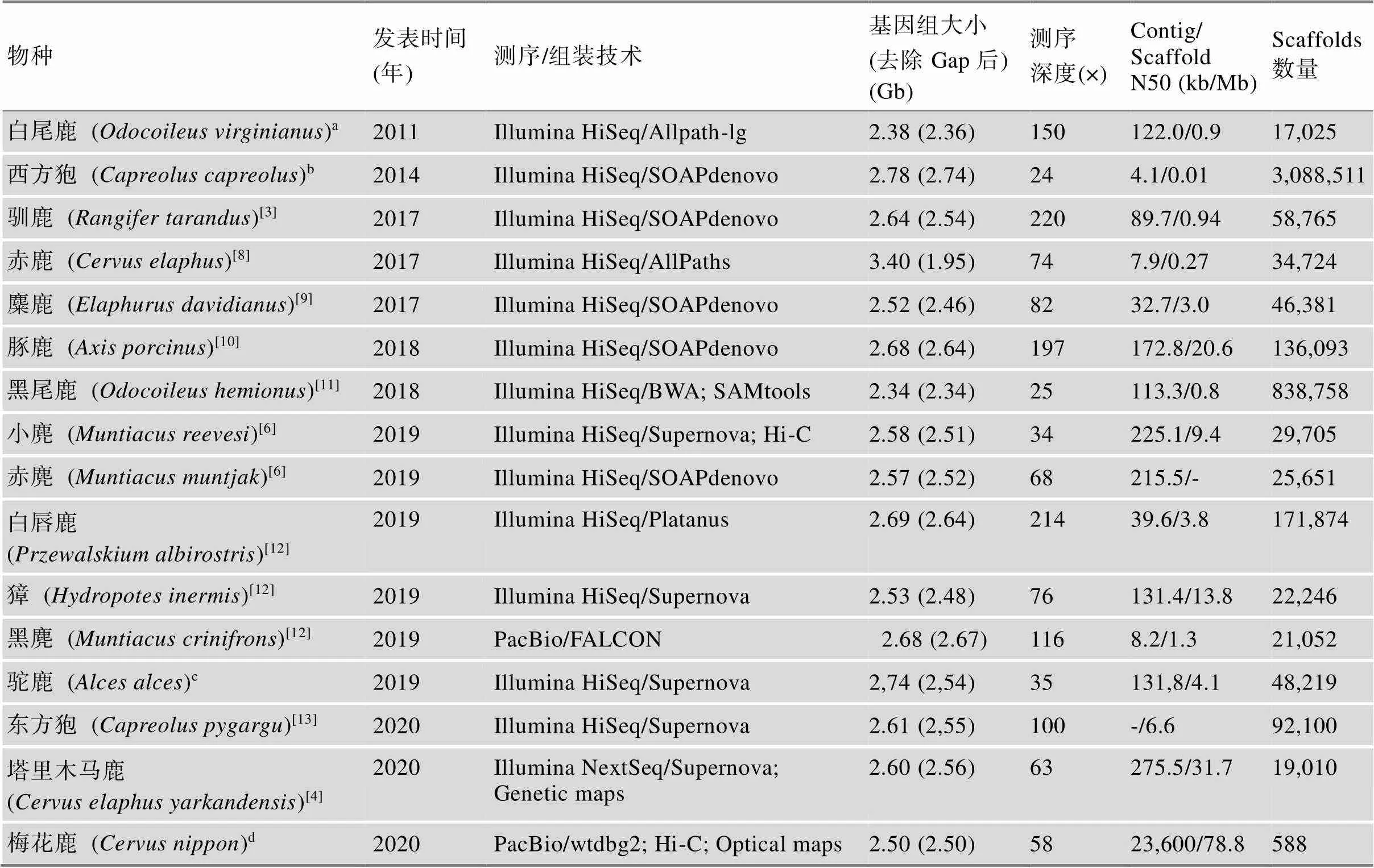
表1 鹿科动物16个参考基因组基本信息
a:GenBank登录号GCA_002102435.1;b:GenBank登录号GCA_000751575.1;c:GenBank 登录号GCA_007570765.1;d:CNGB登录号GWHANOY00000000。
1.2 基因组变异资源
由于鹿科动物参考基因组缺乏,而且对野生动物进行大规模基因组测序显然不太现实。目前,鹿科动物的遗传变异数据主要是基因组SNP位点,其主要来源于简化基因组测序[11,19~23]、牛和羊SNP芯片跨科物种分型[24,25]以及保守牛外显子目标测序[26]和低覆盖率重测序[27,28](表2),这严重制约了鹿科动物基因组变异数据的产出效率和质量。比如,通常简化基因组测序仅产生相当于1%~2%的基因组的SNP数据[22],另外简化基因组测序短序列聚类时,多拷贝重复序列容易导致SNP位点的假阳性[29]。
在跨科物种SNP分型上,Haynes等[24]利用牛BovineSNP50芯片对北美白尾鹿和黑尾鹿()进行分型,分型成功率仅为38.7%。Kharzinova等[25]利用BovineSNP50和OvineSNP50芯片对驯鹿进行分型,分型成功率也仅为43.0%和47.0%。Shafer等[30]阐述在牛科和鹿科(分化时间大约2~3千万年)之间跨芯片分型是不合适的,不仅分型成功率低,而且多态位点仅为1%。
Brauning等[28]对马鹿/赤鹿类群8只个体基因组进行了低覆盖率重测序,利用牛基因组做参考比对并结合严格的SNP筛选策略,共获得了1.8×105个SNP位点,产生了Illumina CervusSNP50芯片。Rowe等[31]利用CervusSNP50芯片对9个鹿种共396只个体进行SNP分型检测,分型成功率为82.3%,远远高于牛羊芯片的分型成功率。Johnston等[5]利用CervusSNP50芯片构建了马鹿/赤鹿类群的遗传连锁图谱,其标记密度和图谱的完整性远远好于Slate等[32]在2002年发表的仅含有600个标记的鹿遗传图谱。

图1 16个基因组测序鹿种系统进化关系及表型数据
系统进化拓扑关系参考文献[14~16],表型数据参考图书《中国鹿科动物》[1]和《The Biology of Deer》[17],核型数据参考王宗仁等[18],鹿科动物照片来源于Wikimedia共享(https://commons.wikimedia.org/)。体重和角重均为平均值。
我国鹿类动物遗传资源十分丰富,同时也是重要的家养经济动物。Hu等[33]对我国5个主要鹿种(包含21个亚种/群体)进行了简化基因组测序,平均测序深度16×,获得197,543个SNP位点,并进行了遗传多样性和系统进化分析。Ba等[19]对我国东北7个地区饲养的42只梅花鹿纯种个体进行双酶切简化基因组测序,平均测序深度23×,共筛选98,166个SNP位点,平均个体SNP密度为0.74 SNPs/kb,略高于经历过严重群体瓶颈的麋鹿(0.51 SNPs/kb)[34],但低于大熊猫(1.32 SNPs/kb)[35]和荷斯坦奶牛(1.35 SNPs/kb)[36]约2倍,而且出现杂合子缺陷。这些结果表明人们长期以茸重性状为单一育种目标,强烈的人工选择导致梅花鹿饲养群体经历了严重的净化和遗传漂变。Hu等[27]通过基因组重测序对梅花鹿鹿茸高产和低产性状进行关联分析,共获得94个与鹿茸重量相关的候选SNP位点。Hu等[37]进一步利用这94个SNP位点对341只个体进行分型,并结合高、低产鹿茸转录组学基因表达数据[38],筛选出16个SNP位点与茸重性状显著相关。

图2 16个基因组测序鹿种的自然地理分布示意图
数据来源于IUCM Red List网站(https://www.iucnredlist.org/)。
由于不同鹿种之间能够杂交且后代可育,人们为了繁育高产性状的茸鹿,经常在饲养梅花鹿和马鹿之间进行杂交。Ba等[20]对纯种梅花鹿和马鹿及杂交F1共30只个体进行双酶切简化基因组测序,平均测序深度15×,获得2015个梅花鹿和马鹿种特异SNP标签,这些标签为纯种和杂交种鉴定奠定了基础。在鹿种鉴定上,Xie等[39]也通过挖掘鹿科动物基因组中保守且与牛羊比较存在突变位点的编码基因,最终在基因序列上开发出1对鹿科动物PCR通用引物,与鹿种特异引物相比,通用引物节省了更多的人力和物力。
在鹿科动物基因组测序之前,科学家通常利用几个牛微卫星多态(short tandem repeats, STRs)标记对鹿科动物进行分型,由于标记数量少,多态信息严重不足。随着高通量测序技术发展,鹿科动物STRs也被快速批量开发。Jia等[40]在梅花鹿多种组织转录组数据[41]中开发了29个STRs,对140只个体进行茸重性状关联分析,找到8个显著关联的标记。Wang等[42]在梅花鹿基因组数据中开发了29个STRs,其中10个高度多态的STRs被用作梅花鹿亲子鉴定标记,排除准确率达99.99%。总之,鹿科动物遗传变异数据的获得不仅对鹿科学研究极具价值,也能为开发和利用经济价值极高的家养鹿类动物提供强有力的遗传分析和改良工具。
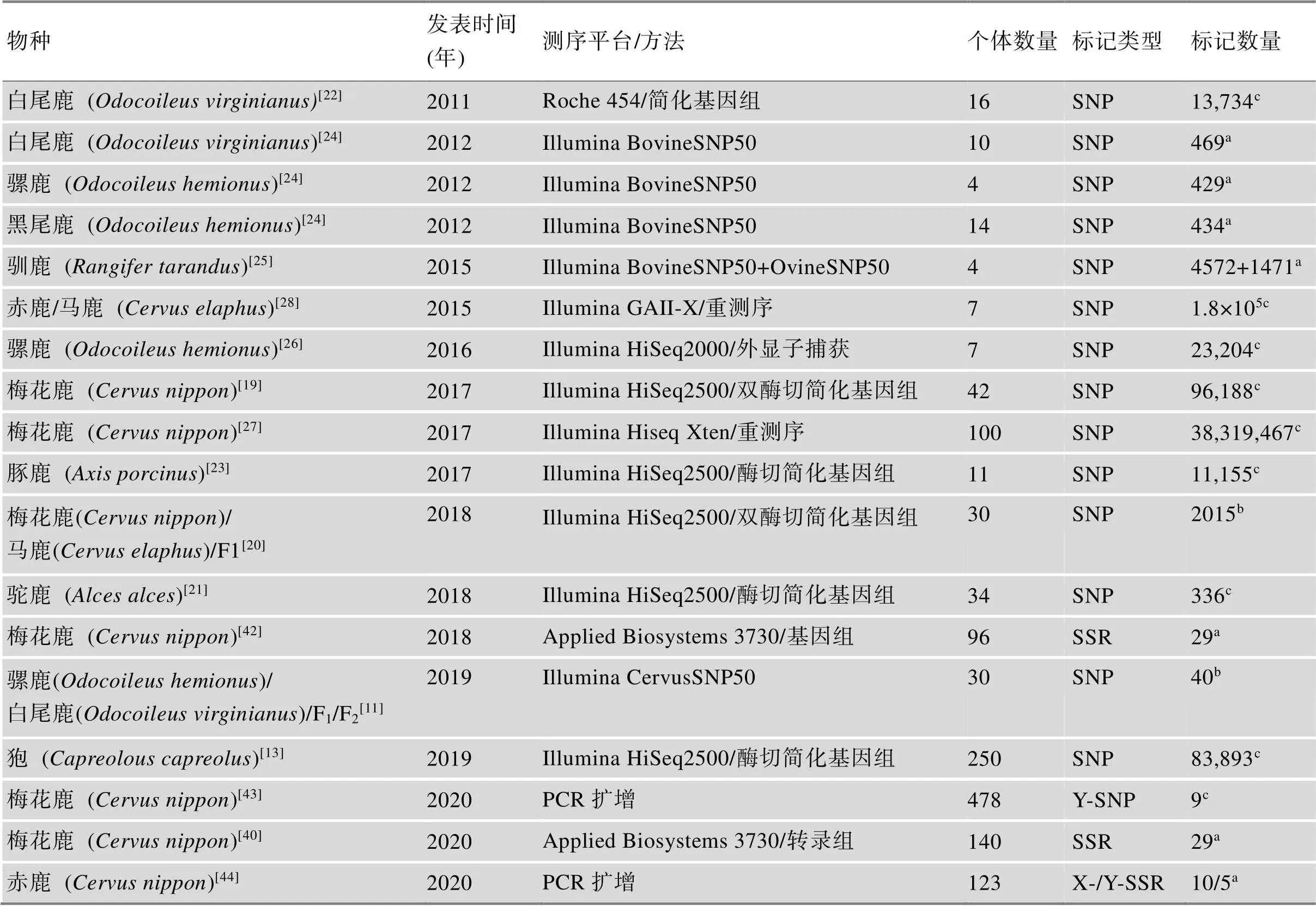
表2 鹿科动物基因组变异资源
a:具有多态性;b:物种特异的SNP;c:最小等位频率至少大于0.05。
2 比较基因组学
2.1 染色体核型进化
染色体重排在驱动进化中的作用一直是进化生物学的长期问题。Farre等[45]重构了反刍动物祖先染色体核型,发现反刍动物祖先与鲸偶蹄目(Cetartiodactyla)分离后染色体重排主要发生在染色体内部,而在反刍亚目(Ruminantia)的有角下目(Pecora)谱系中,染色体重排也发生在染色体之间。反刍动物染色体断点区域附近的基因在物种之间,尤其是在牛中表现出更多的差异表达,并且在具有增强子的基因中差异更大,与这些断点区域的系统发生起源相一致。这些结果表明与染色体重排共定位的谱系特异性调控元件可能提供了有助于塑造反刍动物进化有价值的功能修饰。
在鹿科动物中,麂属动物的核型在较短的分化时间内(仅数百万年)发生了巨大变化。除小麂(2= 46)外,其他麂属动物的染色体数目通常只有6~9条,其中赤麂(2=6♀/7♂)是哺乳动物中染色体数目最少的物种。因此,麂属动物成为研究哺乳动物染色体核型生物学研究的理想材料。通过细菌人工染色体文库的荧光原位杂交建立了麂属动物之间的高分辨比较染色体图谱,确定了麂属动物染色体的串联融合进化方式[46~51]。最近,Mudd 等[6]通过基因组测序和染色体组装,进一步发现小麂和赤麂这两个近缘麂显著的核型差异主要是在基因组上发生了大范围的染色体串联融合事件,而核型急剧变化对局部染色体改变影响甚小,甚至在融合位点附近参与染色体维护的基因很少显示出快速进化的证据。对于不同鹿种的染色体进化,Huang等[52]证实梅花鹿(2= 33)和马鹿(2=34)的染色体核型进化过程中存在唯一的罗伯逊易位。
另外,性染色体起源及演化机制一直是进化生物学家最感兴趣的科学问题之一。Huang等[53]研究发现雄性黑麂基因组中1号常染色体短臂(1p)和4号常染色体发生罗伯逊易位,之后又经历了复杂的内部重排形成年轻新性染色体1p+4。Zhou等[54]研究发现黑麂与哺乳动物Y染色体的演化历程相似,可以成为哺乳动物性染色体演化研究的珍贵模型。关于麂属动物的染色体核型进化,Huang等[55]在2012年进行了详细综述,限于篇幅限制,本文不再综述。
2.2 适应性进化
鹿科动物在北极和热带均有分布,一些鹿种在长期进化中已经适应了极端环境。最近,Lin等[56]揭示了驯鹿适应北极环境分子机制。通过比较基因组发现驯鹿维生素D代谢通路中的两个关键基因(和)受到了强烈的自然选择,基因编码酶的活性比山羊和狍子()高,这可能使驯鹿对钙的吸收能力大大增强。脂蛋白转运()和脂质合成()两个重要基因在驯鹿中也发生了选择性突变,这两个基因也参与了企鹅()和北极熊()脂肪代谢进化,表明极地动物能量代谢经历了趋同进化。驯鹿节律通路中的核心调控基因()发生了驯鹿特异性突变,导致基因与另一个节律核心基因()无法结合,使得驯鹿丧失了昼夜节律分子钟而适应北极极昼和极夜的环境。这些结果使人们对极地动物的适应性在分子水平上有了更深入和全面的了解,也为解决人类一些健康问题提供了重要线索,如维生素D对钙沉积的影响、生物钟调控与人类睡眠障碍的关系。
另外,Weldenegodguad等[57]发现西伯利亚人极地寒冷适应性基因在驯鹿基因组上发生了基因家族扩张和正选择,特别是离子通道受体和离子交换调控基因,如和等,这可能是对温度敏感适应性的主要原因。在类似的研究中,Yang等[58]对不同栖息环境下的脊椎动物重要的冷感受器蛋白TRPM8进行了氨基酸成像与蛋白质三维结构计算建模的研究,发现TRPM8通道的功能结构在南极洲的帝企鹅()与非洲大陆的非洲象()之间存在重大差异,帝企鹅的冷敏感性显著低于非洲象。在鹿科动物中,塔里木马鹿生存于极端干燥、炎热和强烈太阳辐射的沙漠环境。Ababaikeri等[59]通过对塔里木马鹿比较基因组学研究发现该物种的沙漠环境适应性候选基因,主要参与了氧化应激、水分再吸收、能量代谢、热应激、呼吸系统适应、DNA损伤和修复等生物过程。同时,这些候选基因与其他沙漠动物适应性基因均有重叠,表明沙漠物种对沙漠环境适应经历了趋同进化。
麋鹿被认为是急剧恢复和拯救高度濒危动物的经典例子。我国现存麋鹿群体是来自于英国动物园的18只个体繁育后代,这预示着严重的瓶颈效应和近交衰退。Zhu等[34]利用比较基因组学对麋鹿种群的适应性进行了评估,尽管麋鹿群体存在极端的种群瓶颈,但其仍具有较高的遗传多样性,延长的近亲繁殖历史可能有助于清除有害的隐性等位基因,同时,与生殖,胚胎发育和免疫反应相关的17个基因忍受着高选择压力。另外,麋鹿对高盐食物的潜在适应可能是由于基因受到强烈正选择,参与控制体内钠重吸收。另外的29个正选择基因涉及血压调节、心血管发育、胆固醇调节、血糖控制和甲状腺激素合成。这些正选择的基因可能也反应了麋鹿对高盐食物的适应与响应。
鹿科动物进化不仅仅体现在极端环境适应性方面,Ba等[60]发现鹿嗅觉系统适应性进化的分子证据。有51个快速进化基因与感受器官细胞纤毛(cilia)组装有关,同时,实现细胞纤毛波形功能的轴突纤毛丝动力蛋白(dynein)家族重链基因在基因组上发生了扩张,也发现了14个嗅觉受体基因中89个位点在鹿上发生了正选择。由于鹿存在多处皮肤腺体,散发的化学小分子是彼此之间通讯的重要信息素,推测嗅觉系统在接受信息素的长期进化过程中一定也受到了适应性进化的选择压力。
2.3 再生器官(鹿茸角)起源进化关键基因
鹿茸角是哺乳动物中唯一的胚胎组织在成体后发育的器官[61],是鹿科动物防御天敌和争夺配偶的武器。更重要的是,鹿茸角能周期性再生而保持最佳战斗状态,被认为是鹿科动物长期进化过程中一个器官创新。最近,Wang等[14]对反刍动物角起源进化的比较基因组研究发现,鹿茸角新器官的起源依赖招募骨组织、皮肤组织、脑组织和睾丸组织的基因。鹿科动物正选择基因、加速进化元件以及生茸组织高表达基因都参与了神经脊细胞迁移通路,揭示神经脊干细胞在鹿角起源发育中扮演重要角色[62]。Price等[63]也报道了生茸组织特异性高表达神经脊细胞的关键标志物,如等。
鹿科动物中仅有獐在自然条件下是不长茸角的,被认为是在进化过程中发生了茸角丢失[64],因此,利用比较基因组学研究鹿角关键再生基因,獐是唯一的选择。Wang等[14]发现绵羊角的关键调控基因在麝科和獐两个无角反刍动物支系中发生趋同的假基因化,认为也是决定鹿科动物茸角再生的关键基因。Lin等[56]发现驯鹿基因组上促进细胞增殖的细胞周期因子基因上游增加了一个雄性激素受体结合区域,这可能使驯鹿在更低的雄激素水平下能促成雌性驯鹿长角。结果表明,鹿科动物进化过程中,细胞周期相关基因与鹿茸角的发生与再生关联紧密。
总之,比较基因组学是从进化的视角去解析复杂的生物学问题和探寻调控基因。随着多个鹿基因组序列完成,基因组质量也越来越高,将进一步加快鹿科动物比较基因组学研究进程,探索适应性进化和新器官起源的奥秘。
3 功能基因组学
目前,鹿类动物功能基因组学研究主要集中在鹿茸干细胞、鹿茸再生以及鹿茸生长中心的快速生长发育等研究,本文主要针对这几方面进行了总结。
3.1 鹿茸干细胞
鹿茸发生和再生过程是由干细胞驱动的,被称为鹿茸干细胞(antler stem cell, ASC)[2,65,66]。国内外多个实验室已对ASC进行了分离鉴定,其不仅表达多种成体干细胞标记因子,如CD73、CD90、CD29、CD44、CD146、CD105、CD166、STRO-1、Vimentin和Nestin,还表达部分胚胎干细胞标记因子,如C-Myc和OCT4[67~70]。最近,Wang等[69]通过膜蛋白组学发现RXFP2在ASC中高表达,而在对照的面部骨膜细胞中几乎不表达,该蛋白与反刍动物长角有关,可能作为ASC鉴定的一个新标记。在microRNA水平上,Ba等[71]通过测序显示ASC表达小鼠胚胎期特异表达的。Ba等[72]也对培养的ASC进行单细胞RNA-seq测序,表明ASC在培养过程中仍然保持着高度的干性和同质性。Wang等[70]构建胚胎嵌合实验模型证实了ASC不同于胚胎干细胞嵌合到整个宿主的组织中,也不同于间充质干细胞完全消失,而是有限地嵌合到一些组织中,特别是嵌合到了生殖系统中。这些结果证实ASC不仅是成体干细胞,还具有胚胎干细胞部分属性。
干细胞的自我更新、分化与成熟均与其所处的微环境有关。研究发现鹿皮可能为ASC提供了类似的微环境[73]。在鹿茸发生时,雄性激素刺激ASC缓慢增殖和分化,而当分化的组织与头部皮肤变得接触并紧密相贴时,皮肤给ASC提供了特殊的微环境,这个微环境触发了ASC快速增殖(分裂速度比接触前至少快10倍)和定向分化,最终导致鹿茸的发生。类似地,当骨化的鹿角脱落后,伤口开始愈合,ASC再次与皮肤紧密接触并被致敏,ASC再由致敏状态转变成激活状态,启动鹿茸再生。Dong等[74~76]通过蛋白质组学研究表明鹿茸再生的起始阶段是一系列蛋白调控的,可能与上皮-间质转化过程有关,包括无疤痕愈合的第一阶段,源于ASC分化细胞的移动性也受到高度调节,休眠期的ASC和面部骨膜细胞可能使用相似机制来维持干细胞的休眠状态。Sun等[77]通过建立ASC与毛乳头细胞共培养体系,鉴定出128个小分子,60%以上与外泌体相关,信号通路富集分析表明这些分子参与PI3K/AKT信号,可能影响ASC成骨细胞分化和血管生成。Li等[78]利用双向电泳方法对ASC与面部骨膜细胞进行蛋白质组学比较研究,也发现了PI3K/AKT信号在ASC细胞差异表达蛋白中富集。Liu等[79]利用小分子抑制剂影响PI3K/AKT信号的细胞生物学实验进一步证实PI3K/AKT信号在ASC生物学功能中扮演重要角色。
钙网蛋白CALR是一种多功能蛋白,在内质网管腔内主要起钙离子结合蛋白的作用,与多种信号系统有关,例如蛋白质折叠和钙的调节体内平衡。最新证据表明,CALR可在细胞核中与雄激素受体结合,抑制雄激素受体下游转录活动[80]。Dong等[76]通过蛋白质组学研究发现CALR在开始分化的ASC中高表达,并在蛋白互作网络中扮演核心(hub)基因的角色。Akhtar等[81]通过对鹿施以外源雄性激素,也发现雄性激素在低水平时ASC开始分化,高表达CALR,进一步证实了该因子可能是雄激素依赖性鹿茸角再生的主要调节因子。
3.2 鹿茸生长中心
2004年和2012年,研究人员分别利用蛋白质组学和转录组学研究鹿茸生长中心的基因表达。Park等[82]鉴定到了130个蛋白,其中MAPK1COL1A1SMUBP2和ZFP28等仅在鹿茸生长中心高表达,推测这些蛋白对鹿茸的快速生长极其重要。Yao等[83,84]建立了鹿茸生长中心快速生长和骨化的转录组表达谱,涉及转录因子、信号分子和细胞外基质等。鹿茸快速生长发育是基于鹿茸顶端生长中心的ASC增值和分化的。鹿茸发育60天进入快速生长期,90天时开始快速骨化。为了研究不同时间点鹿茸生长发育相关基因表达动态,很多学者都对不同发育时间的鹿茸生长中心进行了转录组和蛋白组分析[83,85~87]。然而,由于鹿茸生长中心组织层结构发育关系复杂,这些研究的样品采集标准不尽相同,难以在不同的研究之间进行有效地比较。
Li等[88]根据发育生物学理论对鹿茸生长中心进行了组织层空间划分,即从远端到近端划分5个连续发育状态的组织层,包括间充质层、前软骨层、过渡层、软骨层和矿化层。基于生长中心组织层发育关系,Ba等[89]对这5个组织层进行了转录组测序,发现不同组织层之间有明显的表达谱特征,而且共表达网络分析鉴定了9个基因表达模块,其中370个核心(hub)基因参与调控了软骨、骨和血管生成,为鹿茸生长中心生长发育研究奠定了分子基础。Ker等[90]建立了鹿茸顶端ASC和人间充质细胞的比较模型,通过转录组测序及体外实验验证阐明对鹿茸细胞快速增殖起主要作用,而是鹿茸矿化的关键基因。Chen等[91]分析lncRNA在鹿茸生长中心间充质组织和软骨组织中的表达动态。与软骨组织相比,间充质组织中有1212个lncRNA和518个mRNA的转录水平发生了显着改变,表明lncRNA通过调节细胞增殖、迁移和成骨相关基因,促进了鹿茸间充质组织向软骨组织的分化。
Hu等[38]对具有快速和慢速两种表型的鹿茸生长中心进行microRNA表达谱比较,发现与鹿茸快速生长的18个microRNA分子,构建microRNA- mRNA调控网络,确定14个基因与鹿茸快速生长相关。Yao等[92]也对鹿茸主干和眉支两个生长中心进行转录组测序,比较发现主干生长中心的软骨发育基因显著上调,如和等,而骨化基因显著下调,如和等。
4 结语与展望
“人类基因组计划”的完成标志着人类对于生命现象和过程有了本质的认识。鹿科动物在长期自然进化过程中,产生了诸多优异性状,如鹿茸角的再生和快速生长[65,93~95]、低癌症发生率[96,97]、无伤疤伤口愈合能力[98,99]、骨质疏松快速逆转[100~102]和大型哺乳动物滞育模型(狍子)[103~107]等。这些都触及了重大基础生物学和医学问题,其背后所蕴藏的遗传机理非常值得人们去认知和掌握。随着测序技术的发展,以单分子荧光和纳米孔为代表的三代测序技术将进一步提升鹿科动物基因组质量,完整的基因组学数据将为这些优异独特的生物学性状相关基因发现奠定了基础。
最近,家养梅花鹿和马鹿也已被纳入我国畜禽遗传资源目录。鹿遗传变异数据大规模产出和利用能有效加快家养鹿类动物的遗传改良和品种保护,比如,利用基因组学数据全面评估我国茸鹿品种遗传结构多样性,利用连锁分析和关联分析等基因组学方法,高效挖掘茸鹿品种中有利等位基因,提出其利用途径和具体方案,并在品种资源创新过程中充分利用基因组学研究成果提高创新效率。
重要的是,针对鹿茸角这一进化过程中独特进化的再生器官,尚有不少重要问题有待解决,如多种细胞类型参与和调控下的鹿茸再生和发育,其它再生模式生物中扮演“总开关”调控角色的免疫细胞是否也扮演了类似的角色。常规组学技术无法详细系统地研究鹿茸角再生和发育过程中细胞类型、发育状态和分化路径。然而,近年来快速发展的单细胞测序技术给发育生物学带来了革命性的变化,能够对单个细胞内基因表达和调控进行无差别的分析,通过更多维、更精准的研究视角,去系统地、全面地揭示和解析细胞的功能。目前,本研究团队正在利用单细胞测序技术绘制鹿茸角再生和发育过程中单细胞图谱,解析不同细胞类型之间互作和通讯以及细胞谱系演化路径等,在单细胞水平上诠释鹿茸干细胞如何再生和发育成完整器官的关键科学问题。未来,可以通过建立以鹿茸为再生医学研究模型开展人类再生医学研究。在实现这些研究目标过程中,鹿基因组学研究将起到不可替代的作用,由此可见,鹿科动物基因组学研究才刚刚开始。
[1] 盛和林. 中国鹿科动物. 生物学通报, 1992, (05): 4–7.
[2] Li CY, Yang FH, Sheppard A. Adult stem cells and mammalian epimorphic regeneration-insights from studying annual renewal of deer antlers., 2009, 4(3): 237–251.
[3] Li ZP, Lin ZS, Ba HX, Chen L, Yang YF, Wang K, Qiu Q, Wang W, Li GY. Draft genome of the reindeer ()., 2017, 6(12): 1–5.
[4] Ba HX, Cai ZX, Gao HY, Qin T, Liu WY, Xie LW, Zhang YL, Jing BY, Wang DT, Li CY. Chromosome- level genome assembly of tarim red deer,., 2020, 7(1): 187.
[5] Johnston SE, Huisman J, Ellis PA, Pemberton JM. A high-density linkage map reveals sexual dimorphism in recombination landscapes in red deer ()., 2017, 7(8): 2859–2870.
[6] Mudd AB, Bredeson JV, Baum R, Hockemeyer D, Rokhsar DS. Analysis of muntjac deer genome and chromatin architecture reveals rapid karyotype evolution., 2020, 3(1): 480.
[7] Ludt CJ, Schroeder W, Rottmann O, Kuehn R. Mitochondrial DNA phylogeography of red deer ()., 2004, 31(3): 1064– 1083.
[8] Bana NÁ, Nyiri A, Nagy J, Frank K, Nagy T, Stéger V, Schiller M, Lakatos P, Sugár L, Horn P, Barta E, Orosz L. The red deergenome cerela1.0: Sequencing, annotating, genes, and chromosomes., 2018, 293(3): 665–684.
[9] Zhang CZ, Chen L, Zhou Y, Wang K, Chemnick LG, Ryder OA, Wang W, Zhang GJ, Qiu Q. Draft genome of the milu ()., 2018, 7(2).
[10] Wang W, Yan HJ, Chen SY, Li ZZ, Yi J, Niu LL, Deng JP, Chen WG, Pu Y, Jia XB, Qu Y, Chen A, Zhong Y, Yu XM, Pang S, Huang WL, Han Y, Liu GJ, Yu JQ. The sequence and de novo assembly of hog deer genome., 2019, 6: 180305.
[11] Russell T, Cullingham C, Kommadath A, Stothard P, Herbst A, Coltman D. Development of a novel mule deer genomic assembly and species-diagnostic snp panel for assessing introgression in mule deer, white-tailed deer, and their interspecific hybrids., 2019, 9(3): 911–919.
[12] Chen L, Qiu Q, Jiang Y, Wang K, Lin ZS, Li ZP, Bibi F, Yang YZ, Wang JH, Nie WH, Su WT, Liu GC, Li QY, Fu WW, Pan XY, Liu C, Yang J, Zhang CZ, Yin Y, Wang Y, Zhao Y, Zhang C, Wang ZK, Qin YL, Liu W, Wang B, Ren YD, Zhang R, Zeng Y, da Fonseca RR, Wei B, Li R, Wan WT, Zhao RP, Zhu WB, Wang YT, Duan SC, Gao Y, Zhang YE, Chen CY, Hvilsom C, Epps CW, Chemnick LG, Dong Y, Mirarab S, Siegismund HR, Ryder OA, Gilbert MTP, Lewin HA, Zhang GJ, Heller R, Wang W. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits., 2019, 364(6446): eaav6202.
[13] de Jong MJ, Li ZP, Qin YL, Quéméré E, Baker K, Wang W, Hoelzel AR. Demography and adaptation promoting evolutionary transitions in a mammalian genus that diversified during the pleistocene., 2020, 29(15): 2777–2792.
[14] Wang Y, Zhang CZ, Wang NN, Li ZP, Heller R, Liu R, Zhao Y, Han JG, Pan XY, Zheng ZQ, Dai XQ, Chen C, Dou M, Peng SJ, Chen XQ, Liu J, Li M, Wang K, Liu C, Lin ZS, Chen L, Hao F, Zhu WB, Song CC, Zhao C, Zheng CL, Wang JM, Hu WS, Li CY, Yang H, Jiang L, Li GY, Liu MJ, Sonstegard TS, Zhang GJ, Jiang Y, Wang W, Qiu Q. Genetic basis of ruminant headgear and rapid antler regeneration., 2019, 364(6446): eaav6335.
[15] Hassanin A, Delsuc F, Ropiquet A, Hammer C, Jansen van Vuuren B, Matthee C, Ruiz-Garcia M, Catzeflis F, Areskoug V, Nguyen TT, Couloux A. Pattern and timing of diversification of(mammalia, laurasiatheria), as revealed by a comprehensive analysis of mitochondrial genomes., 2012, 335(1): 32–50.
[16] Pitra C, Fickel J, Meijaard E, Groves PC. Evolution and phylogeny of old world deer., 2004, 33(3): 880–895.
[17] Leberg P, Smith MH, Brisbin IL. The biology of deer. Springer New York, 1992.
[18] 王宗仁, 杜若甫. 鹿科动物的染色体组型及其进化. 动物学报, 1983, (03): 214–222.
[19] Ba HX, Jia BY, Wang GW, Yang YF, Kedem G, Li CY. Genome-wide snp discovery and analysis of genetic diversity in farmed sika deer () in northeast china using double-digest restriction site-associated DNA sequencing., 2017, 7(9): 3169–3176.
[20] Ba HX, Li ZP, Yang YF, Li CY. Development of diagnostic snp markers to monitor hybridization between sika deer () and wapiti ()., 2018, 8(7): 2173–2179.
[21] Blåhed IM, Königsson H, Ericsson G, Spong G. Discovery of snps for individual identification by reduced representation sequencing of moose ()., 2018, 13(5): e0197364.
[22] Seabury CM, Bhattarai EK, Taylor JF, Viswanathan GG, Cooper SM, Davis DS, Dowd SE, Lockwood ML, Seabury PM. Genome-wide polymorphism and comparative analyses in the white-tailed deer (): A model for conservation genomics., 2011, 6(1): e15811.
[23] Wang W, Yan HJ, Yu JQ, Yi J, Qu Y, Fu MZ, Chen A, Tang H, Niu LL. Discovery of genome-widesnps by rad-seqand the genetic diversity of captive hog deer ()., 2017, 12(3): e0174299.
[24] Haynes GD, Latch EK. Identification of novel single nucleotide polymorphisms (snps) in deer () using the bovinesnp50 beadchip., 2012, 7(5): e36536.
[25] Kharzinova VR, Sermyagin AA, Gladyr EA, Okhlopkov IM, Brem G, Zinovieva NA. A study of applicability of snp chips developed for bovine and ovine species to whole-genome analysis of reindeer rangifer tarandus., 2015, 106(6): 758–761.
[26] Powell JH, Amish SJ, Haynes GD, Luikart G, Latch EK. Candidate adaptive genes associated with lineage divergence: Identifying snps via next-generation targeted resequencing in mule deer ()., 2016, 16(5): 1165–1172.
[27] Hu PF, Xu JP, Ai C, Shao XJ, Wang HL, Dong YM, Cui XZ, Yang FH, Xing XM. Screening weight related genes of velvet antlers by whole genome re-sequencing., 2017, 39(11): 1090–1101.胡鹏飞, 徐佳萍, 艾成, 邵秀娟, 王洪亮, 董依萌, 崔学哲, 杨福合, 邢秀梅. 利用全基因组重测序分析鹿茸重量相关基因. 遗传, 2017, 39(11): 1090–1101.
[28] Brauning R, Fisher PJ, McCulloch AF, Smithies RJ, Ward JF, Bixley MJ, Lawley CT, Rowe SJ, McEwan JC. Utilization of high throughput genome sequencing technology for large scale single nucleotide polymerphism discovery in red deer and canadian elk., 2015: 027318.
[29] Davey JW, Cezard T, Fuentes-Utrilla P, Eland C, Gharbi K, Blaxter ML. Special features of rad sequencing data: Implications for genotyping., 2013, 22(11): 3151–3164.
[30] Shafer ABA, Miller JM, Kardos M. Cross-species application of snp chips is not suitable for identifying runs of homozygosity., 2016, 107(2): 193–195.
[31] Rowe SJ, Clarke SM, van Stijn TC, Hyndaman DL, ward JF, Km M, Dodds KG, Mcewan JC, Newman S-AN, GW A. Brief communication: Developing genomic tools in the new zealand deer industry., 2015(75): 91–93.
[32] Slate J, Van Stijn TC, Anderson RM, McEwan KM, Maqbool NJ, Mathias HC, Bixley MJ, Stevens DR, Molenaar AJ, Beever JE, Galloway SM, Tate ML. A deer (subfamily) genetic linkage map and the evolution of ruminant genomes., 2002, 160(4): 1587–1597.
[33] Hu PF, Shao Y,C Xu JP, Wang TJ, Li YQ, Liu HM, Rong M, Su WL, Chen BX, Cui SH, Cui XZ, Yang FH, Tamate H, Xing XM. Genome-wide study on genetic diversity and phylogeny of five species in the genus cervus., 2019, 20(1): 384.
[34] Zhu LF, Deng C, Zhao X, Ding JJ, Huang HS, Zhu SL, Wang ZW, Qin SS, Ding YH, Lu GQ, Yang ZS. Endangered père david’s deer genome provides insights into population recovering., 2018, 11(10): 2040–2053.
[35] Zhao SC, Zheng PP, Dong SS, Zhan XJ, Wu Q, Guo XS, Hu YB, He WM, Zhang SN, Fan W, Zhu LF, Li D, Zhang XM, Chen Q, Zhang HM, Zhang ZH, Jin XL, Zhang J, Yang HM, Wang J, Wang J, Wei FW. Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation., 2013, 45(1): 67–71.
[36] Szyda J, Fraszczak M, Mielczarek M, Giannico R, Minozzi G, Nicolazzi EL, Kamiński S, Wojdak- Maksymiec K. The assessment of inter-individual variation of whole-genome DNA sequence in 32 cows., 2015, 26(11–12): 658–665.
[37] Hu PF, Deng YY, Ba HX, Li CY. Association analysis of thirty-one single nucleotide polymorphisms with antler weight in sika deer.2020, 51(6): 990–991.
[38] Hu PF, Wang TJ, Liu HM, Xu JP, Wang L, Zhao P, Xing XM. Full-length transcriptome and microrna sequencing reveal the specific gene-regulation network of velvet antler in sika deer with extremely different velvet antler weight., 2019, 294(2): 431–443.
[39] Xie LW, Deng YY, Shao XQ, Hu PF, Zhao DW, Li CY, Ba HX. Design of a universal primer pair for the identification of deer species., 2020, 13(1): 9–12.
[40] Jia BY, Wang GW, Zheng JJ, Yang WY, Chang SZ, Zhang JL, Liu Y, Li QN, Ge CX, Chen G, Liu DD, Yang FH. Development of novel est microsatellite markers for genetic diversity analysis and correlation analysis of velvet antler growth characteristics in sika deer., 2020, 157(1): 24.
[41] Jia BY, Ba HX, Wang GW, Yang Y, Cui XZ, Peng YH, Zheng JJ, Xing XM, Yang FH. Transcriptome analysis of sika deer in china., 2016, 291(5): 1941–1953.
[42] Yang WY, Zheng JJ, Jia BY, Wei HJ, Wang GW, Yang FH. Isolation of novel microsatellite markers and their application for genetic diversity and parentage analyses in sika deer., 2018, 643: 68–73.
[43] Tanaka K, Hoshi A, Nojima R, Suzuki K, Takiguchi H, Takatsuki S, Takizawa T, Hosoi E, Tamate HB, Hayashida M, Anezaki T, Fukue Y, Minami M. Genetic variation in y-chromosome genes of sika deer () in japan., 2020, 37(5): 411–416.
[44] Frank K, Bana NÁ, Bleier N, Sugár L, Nagy J, Wilhelm J, Kálmán Z, Barta E, Orosz L, Horn P, Stéger V. Mining the red deer genome (cerela1.0) to develop x-and y-chromosome-linked str markers., 2020, 15(11): e0242506.
[45] Farré M, Kim J, Proskuryakova AA, Zhang Y, Kulemzina AI, Li QY, Zhou Y, Xiong YQ, Johnson JL, Perelman PL, Johnson WE, Warren WC, Kukekova AV, Zhang GJ, O'Brien SJ, Ryder OA, Graphodatsky AS, Ma J, Lewin HA, Larkin DM. Evolution of gene regulation in ruminants differs between evolutionary breakpoint regions and homologous synteny blocks., 2019, 29(4): 576–589.
[46] Zhou Q, Huang L, Zhang JG, Zhao XY, Zhang QP, Song F, Chi JX, Yang FT, Wang W. Comparative genomic analysis links karyotypic evolution with genomic evolution in the indian muntjac ()., 2006, 115(6): 427–436.
[47] Lee C, Sasi R, Lin CC. Interstitial localization of telomeric DNA sequences in the indian muntjac chromosomes: Further evidence for tandem chromosome fusions in the karyotypic evolution of the asian muntjacs., 1993, 63(3): 156–159.
[48] Yang F, Carter NP, Shi L, Ferguson-Smith MA. A comparative study of karyotypes of muntjacs by chromosome painting., 1995, 103(9): 642–652.
[49] Yang F, O'Brien PC, Wienberg J, Ferguson-Smith MA. Evolution of the black muntjac () karyotype revealed by comparative chromosome painting., 1997, 76(3–4): 159–163.
[50] Yang F, O'Brien PC, Wienberg J, Neitzel H, Lin CC, Ferguson-Smith MA. Chromosomal evolution of the chinese muntjac ()., 1997, 106(1): 37–43.
[51] Tsipouri V, Schueler MG, Hu S, Program NCS, Dutra A, Pak E, Riethman H, Green ED. Comparative sequence analyses reveal sites of ancestral chromosomal fusions in the indian muntjac genome., 2008, 9(10): R155.
[52] Huang L, Chi J, Nie WH, Wang JH, Yang FT. Phylogenomics of several deer species revealed by comparative chromosome painting with chinese muntjac paints., 2006, 127(1–3): 25–33.
[53] Huang L, Chi JX, Wang JH, Nie WH, Su W, Yang FT. High-density comparative bac mapping in the black muntjac (): Molecular cytogenetic dissection of the origin of mcr 1p+4 in the x1x2y1y2y3 sex chromosome system., 2006, 87(5): 608– 615.
[54] Zhou Q, Wang J, Huang L, Nie WH, Wang JH, Liu Y, Zhao XY, Yang FT, Wang W. Neo-sex chromosomes in the black muntjac recapitulate incipient evolution of mammalian sex chromosomes., 2008, 9(6): R98.
[55] Huang L, Jing MD, Zhou Q, Yang FT, Wang W. Research advances in genome evolution of muntjacs ()., 2012,42(2): 87–95.黄玲, 靖美东, 周琦, 杨凤堂, 王文. 鹿科麂属动物基因组演化研究进展. 中国科学:生命科学, 2012, 42(2): 87–95.
[56] Lin ZS, Chen L, Chen XQ, Zhong YB, Yang Y, Xia WH, Liu C, Zhu WB, Wang H, Yan BY, Yang YF, Liu X, Sternang Kvie K, Røed KH, Wang K, Xiao WH, Wei HJ, Li GY, Heller R, Gilbert MTP, Qiu Q, Wang W, Li ZP. Biological adaptations in the arctic cervid, the reindeer ()., 2019, 364(6446): eaav6312.
[57] Weldenegodguad M, Pokharel K, Ming Y, Honkatukia M, Peippo J, Reilas T, Røed KH, Kantanen J. Genome sequence and comparative analysis of reindeer () in northern eurasia., 2020, 10(1): 8980.
[58] Yang SL, Lu XC, Wang YF, Xu LZ, Chen XY, Yang F, Lai R. A paradigm of thermal adaptation in penguins and elephants by tuning cold activation in trpm8., 2020, 117(15): 8633–8638.
[59] Ababaikeri B, Abduriyim S, Tohetahong Y, Mamat T, Ahmat A, Halik M. Whole-genome sequencing of tarim red deer () reveals demographic history and adaptations to an arid-desert environment., 2020, 17(1): 31.
[60] Ba HX, Qin T, Cai ZX, Liu WY, Li CY. Molecular evidence for adaptive evolution of olfactory-related genes in cervids., 2020, 42(4): 355–360.
[61] Li CY, Suttie JM. Deer antlerogenic periosteum: A piece of postnatally retained embryonic tissue?, 2001, 204(5): 375–388.
[62] Kierdorf U, Li CY, Price JS. Improbable appendages: Deer antler renewal as a unique case of mammalian regeneration., 2009, 20(5): 535– 542.
[63] Price JS, Allen S, Faucheux C, Althnaian T, Mount JG. Deer antlers: A zoological curiosity or the key to understanding organ regeneration in mammals?, 2005, 207(5): 603–618.
[64] Randi E, Mucci N, Pierpaoli M, Douzery E. New phylogenetic perspectives on the() are provided by the mitochondrial cytochrome b gene., 1998, 265(1398): 793–801.
[65] Li CY. Deer antler regeneration: A stem cell-based epimorphic process., 2012, 96(1): 51–62.
[66] Li CY, Chu WH. The regenerating antler blastema: The derivative of stem cells resident in a pedicle stump., 2016, 21: 455–467.
[67] Rolf HJ, Kierdorf U, Kierdorf H, Schulz J, Seymour N, Schliephake H, Napp J, Niebert S, Wölfel H, Wiese KG. Localization and characterization of stro-1 cells in the deer pedicle and regenerating antler., 2008, 3(4): e2064.
[68] Seo MS, Park SB, Choi SW, Kim JJ, Kim HS, Kang KS. Isolation and characterization of antler-derived multipotent stem cells., 2014, 23(7): 831–843.
[69] Wang DT, Ba HX, Li CG, Zhao QM, Li CY. Proteomic analysis of plasma membrane proteins of antler stem cells using label-free LC-MS/MS., 2018, 19(11): 3477.
[70] Wang DT, Berg DB, Ba HX, Sun HM, Wang Z, Li CY. Deer antler stem cells are a novel type of cells that sustain full regeneration of a mammalian organ-deer antler., 2019, 10(6): 443.
[71] Ba HX, Wang DT, Li CY. Microrna profiling of antler stem cells in potentiated and dormant states and their potential roles in antler regeneration., 2016, 291(2): 943–955.
[72] Ba HX, Wang DT, Wu WY, Sun HM, Li CY. Single-cell transcriptome provides novel insights into antler stem cells, a cell type capable of mammalian organ regeneration., 2019, 19(4): 555–564.
[73] Li CY, Yang FH, Li GY, Gao XH, Xing XM, Wei HJ, Deng XM, Clark DE. Antler regeneration: A dependent process of stem tissue primed via interaction with its enveloping skin., 2007, 307(2): 95–105.
[74] Dong Z, Ba HX, Zhang W, Coates D, Li CY. Itraq-based quantitative proteomic analysis of the potentiated and dormant antler stem cells., 2016, 17(11): 1778.
[75] Dong Z, Coates D, Liu QX, Sun HM, Li CY. Quantitative proteomic analysis of deer antler stem cells as a model of mammalian organ regeneration., 2019, 195: 98–113.
[76] Dong Z, Haines S, Coates D. Proteomic profiling of stem cell tissues during regeneration of deer antler: A model of mammalian organ regeneration., 2020, 19(4): 1760–1775.
[77] Sun HM, Sui ZG, Wang DT, Ba HX, Zhao HP, Zhang LH, Li CY. Identification of interactive molecules between antler stem cells and dermal papilla cells using anco-culture system., 2020, 51(1): 15–31.
[78] Li CY, Harper A, Puddick J, Wang WY, McMahon C. Proteomes and signalling pathways of antler stem cells., 2012, 7(1): e30026.
[79] Liu Z, Zhao HP, Wang D, McMahon C, Li CY. Differential effects of the pi3k/akt pathway on antler stem cells for generation and regeneration of antlers., 2018, 23: 1848– 1863.
[80] Dedhar S, Rennie PS, Shago M, Hagesteijn CY, Yang H, Filmus J, Hawley RG, Bruchovsky N, Cheng H, Matusik RJ. Inhibition of nuclear hormone receptor activity by calreticulin., 1994, 367(6462): 480–483.
[81] Akhtar RW, Liu Z, Wang DT, Ba HX, Shah SAH, Li CY. Identification of proteins that mediate the role of androgens in antler regeneration using label free proteomics in sika deer ()., 2019, 283: 113235.
[82] Park HJ, Lee DH, Park SG, Lee SC, Cho S, Kim HK, Kim JJ, Bae H, Park BC. Proteome analysis of red deer antlers., 2004, 4(11): 3642–3653.
[83] Yao BJ, Zhao Y, Wang Q, Zhang M, Liu MC, Liu HL, Li J. De novo characterization of the antler tip of chinese sika deer transcriptome and analysis of gene expression related to rapid growth., 2012, 364(1–2): 93–100.
[84] Yao BJ, Zhao Y, Zhang HS, Zhang M, Liu MC, Liu HL, Li J. Sequencing and de novo analysis of the chinese sika deer antler-tip transcriptome during the ossification stage using illumina rna-seq technology., 2012, 34(5): 813–822.
[85] Zhao Y, Yao BJ, Zhang M, Wang SM, Zhang H, Xiao W. Comparative analysis of differentially expressed genes in sika deer antler at different stages., 2013, 40(2): 1665–1676.
[86] Han RB, Han L, Wang SN, Li HP. Whole transcriptome analysis of mesenchyme tissue in sika deer antler revealed the cernas regulatory network associated with antler development., 2019, 10: 1403.
[87] Zhang YY, Liu HM, Shao YC, Zhou PY, Su Y, Wang L, Xing XM. Comparative proteomic analysis in different growth stages of sika deer velvet antler,, 2016, 47(3): 493–501.张然然, 刘华淼, 邵元臣, 周盼伊, 苏莹, 王磊, 邢秀梅. 不同生长时期梅花鹿鹿茸差异蛋白质组学分析. 畜牧兽医学报, 2016, 47(3): 493–501.
[88] Li CY, Clark DE, Lord EA, Stanton JA, Suttie JM. Sampling technique to discriminate the different tissue layers of growing antler tips for gene discovery., 2002, 268(2): 125–130.
[89] Ba HX, Wang DT, Yau TO, Shang YD, Li CY. Transcriptomic analysis of different tissue layers in antler growth center in sika deer ()., 2019, 20(1): 173.
[90] Ker DFE, Wang D, Sharma R, Zhang B, Passarelli B, Neff N, Li CY, Maloney W, Quake S, Yang YP. Identifying deer antler uhrf1 proliferation and s100a10 mineralization genes using comparative rna-seq., 2018, 9(1): 292.
[91] Chen DY, Jiang RF, Li YJ, Liu MX, Wu L, Hu W. Screening and functional identification of lncrnas in antler mesenchymal and cartilage tissues using high- throughput sequencing., 2020, 10(1): 9492.
[92] Yao BJ, Wang CN, Zhou ZW, Zhang M, Zhao DQ, Bai XY, Leng XY. Comparative transcriptome analysis of the main beam and brow tine of sika deer antler provides insights into the molecular control of rapid antler growth., 2020, 25: 42.
[93] Li CY, Zhao HP, Liu Z, McMahon C. Deer antler--a novel model for studying organ regeneration in mammals.2014, 56: 111–122.
[94] Landete-Castillejos T, Kierdorf H, Gomez S, Luna S, García AJ, Cappelli J, Pérez-Serrano M, Pérez-Barbería J, Gallego L, Kierdorf U. Antlers-evolution, development, structure, composition, and biomechanics of an outstanding type of bone., 2019, 128: 115046.
[95] Fennessy PF. Deer antlers: Regeneration, function and evolution., 1984, 14(3): 290–291.
[96] Lombard LS, Witte EJ. Frequency and types of tumors in mammals and birds of the philadelphia zoological garden.,1959, 19(2): 127–141.
[97] Griner LA. A review of necropsies conducted over a fourteen-year period at the san diego zoo and san diego wild animal park. In: Pathology of Zoo Animals. 1983.
[98] Rong XL, Chu WH, Zhang HY, Wang YS, Qi XY, Zhang GK, Wang YM, Li CY. Antler stem cell- conditioned medium stimulates regenerative wound healing in rats.2019, 10(1): 326.
[99] Willyard C. Unlocking the secrets of scar-free skin healing., 2018, 563(7732): S86–S88.
[100] Banks WJ, Epling GP, Kainer RA, Davis RW. Antler growth and osteoporosis. I. Morphological and morphometric changes in the costal compacta during the antler growth cycle., 1968, 162(4): 387–397.
[101] Jr WJB, Epling GP, Kainer RA, Davis RW. Antler growth and osteoporosis. II. Gravimetric and chemical changes in the costal compacta during the antler growth cycle., 1968, 162(4): 399–405.
[102] Stéger V, Molnár A, Borsy A, Gyurján I, Szabolcsi Z, Dancs G, Molnár J, Papp P, Nagy J, Puskás L, Barta E, Zomborszky Z, Horn P, Podani J, Semsey S, Lakatos P, Orosz L. Antler development and coupled osteoporosis in the skeleton of red deer cervus elaphus: Expression dynamics for regulatory and effector genes., 2010, 284(4): 273–287.
[103] van der Weijden VA, Ulbrich SE. Embryonic diapause in roe deer: A model to unravel embryo-maternal communication during pre-implantation development in wildlife and livestock species., 2020, 158: 105–111.
[104] Beyes M, Nause N, Bleyer M, Kaup FJ, Neumann S. Description of post-implantation embryonic stages in european roe deer (capreolus capreolus) after embryonic diapause., 2017, 46(6): 582–591.
[105] Drews B, Rudolf Vegas A, van der Weijden VA, Milojevic V, Hankele AK, Schuler G, Ulbrich SE. Do ovarian steroid hormones control the resumption of embryonic growth following the period of diapause in roe deer ()?, 2019, 19(2): 149–157.
[106] Korzekwa AJ, Kotlarczyk AM, Zadroga A. Profiles of maternal origin factors during transition from embryonic diapause to implantation in roe deer., 2019, 90(11): 1444–1452.
[107] van der Weijden VA, Puntar B, Rudolf Vegas A, Milojevic V, Schanzenbach CI, Kowalewski MP, Drews B, Ulbrich SE. Endometrial luminal epithelial cells sense embryo elongation in the roe deer independent of interferon-tau†., 2019, 101(5): 882–892.
Progress on deer genome research
Hengxing Ba1,2, Pengfei Hu1,2, Chunyi Li1,2
Deer family is one of the most abundant mammalian families in the world. Deer species are distributed in wide geographic ranges including the North Pole, tropical regions and high-altitude mountains. Of these deer species, China accounts for more than 40% of them and is the main site for deer evolution. Besides the common phenotypical attributes for ruminants, deer family is evolved to possess the unique head gears with periodic regeneration, i.e. antlers. It is currently well accepted that deer is a very valuable model for the studies of ecology, behavior, evolution and biology, especially for the study of mammalian organ regeneration. Reference deer genome is the basis for systematically illustrating deer evolution, deciphering unique biological attributes of deer species, and is significant in protection and utilization of deer genetic resources. In this review, we summarize the recent progress in the field of deer genome research, including data of deer genetic variation, molecular basis of adaptive evolution, and key genes and functional genomics involved in deer antler origin and evolution. The overall aim of the paper is to provide the reference neccessary for in depth investigation of deer species.
cervids; deer genome; genetic variation; adaptive evolution; velvet antler; functional genomic
2020-10-28;
2020-12-29
国家自然科学基金项目(编号:31402035, U20A20403) 和吉林省科技发展计划项目(编号:20200602013ZP)资助[Supported by the National Natural Science Foundation of China (Nos. 31402035, U20A20403), and the Science and Technology Development Project of Jilin Province (No. 20200602013ZP)]
巴恒星,博士,研究员,研究方向:特种动物基因组学。E-mail: bahengxing@caas.cn
李春义,博士,研究员,研究方向:鹿茸生物学。E-mail: lichunyi1959@163.com
巴恒星,博士,研究员,研究方向:特种动物基因组学。E-mail: bahengxing@caas.cn
10.16288/j.yczz.20-362
2021/3/11 17:23:28
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20210310.1027.003.html
(责任编委: 姜雨)
猜你喜欢
成都医学院学报(2021年2期)2021-07-19 08:35:28
基层中医药(2021年11期)2021-06-05 06:54:26
科学(2020年2期)2020-08-24 07:56:44
科学之谜(2019年3期)2019-03-28 10:29:44
科学之谜(2018年8期)2018-09-29 11:06:46
中国现代中药(2017年1期)2017-09-21 09:19:05
环球时报(2017-06-27)2017-06-27 09:13:42
恋爱婚姻家庭·养生版(2016年9期)2016-09-07 11:25:01
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:16
特产研究(2015年1期)2015-04-12 06:36:20
