
溶藻弧菌群体基因组学研究
2021-04-22 07:31郑宏源闫琳杨超武雅蓉秦婧靓郝彤宇杨大进郭云昌裴晓燕赵彤言崔玉军
遗传 2021年4期
郑宏源,闫琳,杨超,3,武雅蓉,秦婧靓,郝彤宇,杨大进,郭云昌,裴晓燕,赵彤言,崔玉军
研究报告
溶藻弧菌群体基因组学研究
郑宏源1,闫琳2,杨超1,3,武雅蓉1,秦婧靓1,郝彤宇1,杨大进2,郭云昌2,裴晓燕2,赵彤言1,崔玉军1
1. 军事科学院军事医学研究院微生物流行病研究所,北京 100071 2. 国家食品安全风险评估中心,北京 100022 3. 深圳市疾病预防控制中心,深圳 518055
溶藻弧菌()是一种能够对人类以及鱼、虾、贝类等水产品致病的弧菌,给人类健康带来威胁,也给水产养殖业造成巨大的经济损失。目前该物种基于全基因组的遗传多样性和重要遗传元件研究报道较少。本研究对采集自全国4个省份的68株溶藻弧菌进行高通量测序,获得全基因组序列,并结合113株公开发表的全球序列数据,利用fineSTRUCTURE软件、VFDB毒力因子库和CARD、ResFinder耐药数据库,对溶藻弧菌的种群结构和毒力、耐药因子分别进行解析。结果表明:溶藻弧菌可分为谱系1和谱系2。两个谱系在美洲和亚洲均有分布,但欧洲仅分离到谱系1菌株;共鉴定发现12个克隆群,其中一个克隆群内菌株存在跨洋传播现象。该物种携带、、等多种不同功能的毒力因子;毒力因子在两个谱系间的分布无特异性,但存在地域间差异:其中欧洲菌株携带、、和的比率低于其他地区,而基因的携带率则明显高于其他地区,我国广西菌株中基因携带率低于其他省份,且不携带。多个基因组携带、、、、、和等与多种抗生素耐受相关的基因,其中和基因在谱系2中的出现频率远高于谱系1;此外,基因在亚洲菌株中的携带率高于美洲和欧洲地区,而在我国四川菌株中的携带率则低于其他省份。在5个基因组中(VA24、VA28、2014V-1011、ZJ-T和Vb1833)观察到质粒或ICE等携带多种耐药基因的大片段。本研究通过群体基因组学的研究方法,揭示了溶藻弧菌的种群结构组成和毒力、耐药相关元件的分布,为进一步了解溶藻弧菌的遗传特征和致病机制提供必要基础,为该病原的监测、预防和控制工作提供科学支撑。
溶藻弧菌;全基因组测序;种群结构;毒力因子;耐药基因
溶藻弧菌(),是一种广泛存在于海洋和河口,可以从病人体内、海水、水产品等多种不同环境中分离得到的革兰氏阴性杆菌[1,2]。溶藻弧菌感染人体后可以引起皮肤感染、外耳道感染等多种疾病,但与霍乱弧菌()和副溶血弧菌()等食源性弧菌不同,临床上报导的溶藻弧菌感染病例均有海水暴露史,因此其主要感染途径是与海水的接触[3,4]。此外,溶藻弧菌可引起鱼、虾、贝类等水产品患病与大量死亡,带来巨大的经济损失[2,5,6]。美国COVIS (Cholera and OtherIllness Surveillance)和FoodNet (Foodborne Diseases Active Surveillance Network)两个监测数据库的数据表明:自2007年以来,溶藻弧菌已经跃升为第二常见的致病弧菌[7,8],为公共卫生及渔业经济带来巨大压力。深入研究溶藻弧菌遗传多样性和病原相关特征,将有助于该病原的精准检测和致病机制研究,对其监测和防控工作具有重要意义。
多年来,已有多种传统分子生物学方法应用于溶藻弧菌分型研究,例如核糖体分型、随机扩增多态性DNA (random amplified polymorphic DNA, RAPD)、肠杆菌基因间重复一致序列PCR (enterobacterial repetitive intergenic consensus PCR, ERIC- PCR)、低频限制性位点PCR (infrequent restriction site PCR, IRS-PCR)和脉冲场凝胶电泳(pulsed field gel electrophoresis, PFGE)等[9~12]。这些方法在溯源性研究等方面发挥了一定的作用,具有操作简便、获得结果较快等优势,但缺点是分辨率较低,实验结果不稳定,而且对实验条件要求较高。PFGE曾被认为是暴发溯源的金标准[13,14],但是这种分型方法需要严苛的实验条件,实验过程中操作导致的细微差别则可能得到不同的分型结果,不利于实验结果在不同实验室间的交流。目前尚无多位点可变数量串联重复序列分析(multilocus variable-number tandem repeat analysis, MLVA)和多位点序列分型(multilocus sequence typing, MLST)等多靶标分子分型方法应用于溶藻弧菌的研究报导,而基于全基因组层面的遗传多样性研究和种群结构鉴定的研究报道较少。另一方面,当前对于溶藻弧菌的毒力和耐药相关基因的研究,是利用已知其他弧菌物种的毒力和耐药基因特异性引物,通过PCR方法在部分菌株中检验相应基因是否存在。研究表明溶藻弧菌携带了一些副溶血弧菌等物种常见毒力因子,也表现出多种临床常见抗生素的耐受性,同时鉴定到部分菌株携带有耐药基因[1,15~21]。然而,溶藻弧菌全基因组层面毒力和耐药相关遗传因子分布的认识仍不够深入。
全基因组测序(whole genome sequencing, WGS)在细菌遗传多样性研究中有着最高的分辨率,而且已广泛运用在微生物溯源、传播及种群结构鉴定等各个方面[22~25]。本研究通过全基因组测序,利用fineSTRUCTURE方法[26]鉴定了溶藻弧菌的种群结构,并在此基础上研究了毒力、耐药相关因子及其在物种内不同菌株间的分布。
1 材料与方法
1.1 菌株来源
本研究共收集181株溶藻弧菌的全基因组序列,包括68株新测序的序列和113株公共基因组序列。新测序的菌株采样于国内四个省份(山东、湖北、四川、广西)的水产品、海水、水底沉积物等不同环境,采样时间均为2014年。公共序列下载于NCBI数据库(National Center for Biotechnology Information Search database,https://www.ncbi.nlm.nih.gov/),包括截止到2020年10月15日的所有组装结果和SRA数据(46+67),这些菌株来自美洲、欧洲和亚洲,分离时间为1969−2020年间。
1.2 全基因组测序
使用细菌基因组DNA提取试剂盒(QIAGEN UltraClean®Microbial DNA Isolation kit),参照试剂盒说明书提取全基因组DNA。经检测质量合格后送测序公司(华大基因,深圳)进行细菌全基因组测序,测序平台为Illumina NovaSeq 6000,测序文库大小为350 bp,测序reads为双端150 bp,平均测序深度134X (94~188X)。
1.3 质控及组装
使用软件Trimmomatic v0.38[27]将测序结果中低质量的数据进行过滤,然后利用软件shovill (https:// github.com/tseemann/shovill)对过滤后的reads 进行组装。组装后的基因组草图序列已上传至NCBI,BioProject编号为PRJNA714277。
1.4 变异检测和基因预测
单核苷酸多态性(single nucleotide polymorphism, SNP)位点的鉴定参照我们已发表的方法[28],以溶藻弧菌FA2(GCA_011801435.1)基因组完成图为参考序列,使用MUMmer软件包[29]将181株溶藻弧菌基因组序列分别比对到参考菌株基因组,获得核心基因组(所有菌株中均能找到对应碱基状态)SNP矩阵。为保证SNP鉴定结果的可靠性,位于重复区和测序质量值低的SNP 未纳入后续分析。采用Prokka软件[30]对各个菌株基因组序列进行基因预测,使用Roary软件[31]判断基因获得缺失。
1.5 系统发育分析
使用TreeBest软件(http://treesoft.sourceforge. net/treebest.shtml)对SNP数据集构建邻接(Neighbour- Joining, NJ)树,bootstrap值设为100,使用在线工具iTOL[32]进行可视化展示。
1.6 种群结构鉴定
首先将SNP矩阵转换为FASTA格式,然后计算两两菌株间的SNP距离,为减小克隆结构的干扰,以10,000个SNP为阈值去除克隆信号,并从每个克隆群中随机选取一株菌作为该群的代表株,经过筛选最终得到129个代表株。将129株菌的SNP信息输入种群结构分析软件fineSTRUCTURE[26],设定默认参数,计算菌株间共祖率(coancestry),得到共祖率矩阵,再利用马尔可夫链蒙特卡罗(Markov Chain Monte Carlo, MCMC)算法实施矩阵的聚类分析,进而推断种群结构。
1.7 毒力因子鉴定和耐药基因预测
使用BLASTn将所有菌株基因组数据与VFDB (Virulence Factor Database)[33]数据库核心库(setA,仅包含经试验验证过的毒力基因)进行比对,鉴定本研究中数据集的毒力因子;利用RGI v5.1.1[34]和ResFinder[35]两种软件分别基于CARD (Comprehensive Antibiotic Resistance Database, https://card. mcmaster.ca/)耐药基因库和ResFinder (https://bitbucket. org/genomicepidemiology/resfinder_db/src/master/)耐药库鉴定整个数据集的耐药基因。毒力因子和耐药基因鉴定结果中,参考我们已发表的相关工作[36],coverage和identity值均大于70%认为该基因存在,否则为缺失。
2 结果与分析
2.1 溶藻弧菌基因组特点
本研究在我国4个省份(山东、湖北、四川、广西)共分离了68株溶藻弧菌,加上公共数据113株,共181株菌基因组数据纳入分析。新测序的68株溶藻弧菌基因组序列,经过滤后组装,平均基因组大小为5.1 Mb (4.9~5.4 Mb),平均GC含量为44.6% (44.4%~44.9%)。整个数据集首先进行了泛基因组分析,共得到22,439个基因,其中核心基因2995个,附属基因19,444个。根据图1可以推测,随着菌株数量的增加,整个物种的泛基因数量呈上升趋势,而核心基因组则趋于稳定,说明整个物种的附属基因在逐渐增加,即溶藻弧菌的基因组是“开放”的,可以通过水平基因转移等方式不断获得新的基因。

图1 溶藻弧菌基因数量与基因组数量的关系
横轴为纳入分析的基因组数量,纵轴为基于数据集计算出的基因总数与核心基因数量。
2.2 溶藻弧菌种群结构
通过变异检测,181株溶藻弧菌共鉴定出335,733个SNP。根据鉴定到的SNP,我们构建了整个数据集的系统发育树(图2A),结果显示溶藻弧菌的系统发育树和副溶血弧菌[24,37]等物种类似,呈现“轮盘状”结构(图2A),说明菌株间水平转移信号已将垂直遗传信号打乱,该物种属于高频重组弧菌。使用fineSTRUCTURE软件对溶藻弧菌的种群结构做进一步解析,最终将溶藻弧菌分成两个主要谱系(Lineage):谱系1和2 (图2B)。从图中可发现,在对角线上的少量种群中仍呈现明显的克隆信号,在我们已发表的相关研究中[24]提到,可通过进一步去除克隆群菌株以移除克隆信号干扰。本研究由于菌株数量相对较少,且进一步去除克隆信号后得到的结果仍分为同样两个谱系,故只展示第一次去除克隆信号的结果。
分析结果(表1)显示:谱系1中的美洲和中国菌株占比相当,而谱系2的中国菌株占比较高;谱系1菌株与谱系2菌株相比分布更为广泛,欧洲来源的17株菌仅出现在谱系1,提示谱系1在全球的扩散程度更高。

图2 溶藻弧菌系统发育树与种群结构
A:181株菌基于核心基因组SNP构建的系统发育树。黑色、红色树枝分别代表谱系1和谱系2,树枝顶部紫色圆点代表鉴定到的12个克隆群(clonal groups, CGs),右下角红色虚线框代表的是发生跨洋传播的两株菌,内环代表不同的采样地点,外环代表不同采样来源。B:129株代表菌株基于fineSTRUCTURE软件的种群鉴定结果。列代表供体菌株(donor),行代表受体菌株(recipient),方格颜色表示供体向受体导入的序列片段数量,黑色线代表种群间的界线。

表1 不同谱系中菌株分离地点统计
*代表两个谱系中不同采样地点的菌株数量,#代表相应地点采样菌株在谱系内的占比。
参考我们已发表的研究方法[25],以SNP距离小于10为界鉴定出了近期传播或暴发的12个克隆群(CGs,图2A),共涉及34株菌。仅在其中一个克隆群中(图2A中虚线框),发现来自不同国家的菌株。相关菌株分别来自英国和日本(分离时间不详),这两株菌基因组间 SNP遗传距离为0,表明溶藻弧菌在上述两个国家之间发生了跨地域传播。背景信息显示这两株菌分别分离自蛤类和鱼类,提示跨国传播可能是由于水产品国际贸易导致的。本研究中溶藻弧菌跨洋传播现象的确定对我们将来的防治思路和策略有指导意义。
2.3 毒力因子的鉴定及分布
181个基因组中共鉴定出9类37个毒力因子(表2),其中鞭毛(flagella)与三型分泌系统(T3SS)相关基因最多,分别为14个和16个,占毒力因子总数的81%。鞭毛作为一种附着因子(adhesin factor),可介导溶藻弧菌在宿主表面的粘附;T3SS最初是在耶尔森氏菌属(spp)中发现的[38],而弧菌属(spp.)中最早发现T3SS是在副溶血弧菌(血清型O3:K6, strain RIMD2210633)的两条染色体上,包含两套T3SS系统,分别命名为T3SS1和T3SS2[39],其中T3SS1在所有副溶血弧菌基因组中都存在,主要功能是诱导细胞自噬、细胞圆化和细胞裂解[40,41]。此外,我们还发现所有的溶藻弧菌(100%)都携带溶血素基因。是5类典型溶血素家族(TDH、HlyA、TLH、δ-VPH、HLX)[42]中的一种,实验表明这种基因在宿主中介导了凋亡、膜溶解、坏死等多种毒性表现[42,43]。98%的菌株携带膜外蛋白基因,这是溶藻弧菌的主要孔蛋白,也是一种重要的毒力因子,有研究已经证实在溶藻弧菌的铁代谢平衡中起到了重要作用,而且也是溶藻弧菌重要的疫苗研发靶标[44,45]。我们还分别在87%、26%和9%的溶藻弧菌中发现携带多价粘附分子基因、免疫原性脂蛋白基因、和热休克蛋白基因。另有个别菌株表达荚膜基因和T6SS亚单位。

表2 溶藻弧菌毒力因子及相关功能
括号内百分比代表整个数据集中携带此毒力基因的菌株的百分比。
在鉴定出的37个毒力因子中,其中23个在大于90%的菌株中携带, 8个在10%~90%的菌株中携带,其余6个仅在少数(<10%)菌株中携带。统计这8个分布差异性较大的毒力因子在不同谱系中、不同国家和地区间、中国不同省份的分布情况(表3),结合毒力因子在系统发育树上的分布(图3),未发现谱系1或谱系2的群特异性毒力因子;欧洲菌株中、、和的携带率低于其他国家和地区,而基因的携带率较高;除中国外的亚洲其他地区的菌株均不携带基因;此外,我国广西的菌株基因的携带率低于其他省份,且均不携带基因。因本研究中的菌株采样量较小,相关遗传因子的地域分布特征仍需将来通过扩大菌株集进行验证。
2.4 耐药基因的预测及分布
RGI和ResFinder均鉴定到32个耐药基因,经同源比对、去除冗余后汇总为31个耐药基因,分别介导溶藻弧菌对β内酰胺酶类、喹诺酮类、四环素类等多种常见抗生素的耐受(表4)。我们发现几乎所有菌株(>95%)普遍携带的耐药基因有:介导β内酰胺酶类耐受的,介导四环素耐受的和,介导喹诺酮类耐受的,以及调控耐药的基因、。
此外,我们发现介导四环素耐受的基因在两个谱系中的分布存在显著差异(图3),谱系2和谱系1的菌株携带这种基因的比率分别为100%和12%。是一种转录调节因子,与四环素耐药相关,而且另一个四环素耐药基因耐药功能的正常发挥,依赖于。我们还在9株溶藻弧菌中首次发现了一种和磷霉素耐受相关的基因,有趣的是其中8株菌(88.9%)属于谱系2。此外,经统计发现,基因在中国和亚洲其他地区的菌株中的携带率(分别为45%和44%)高于美洲和欧洲菌株的携带率(分别为24%和6%);而我国四川菌株中该基因的携带率(14%)低于其他省(广西:60%,山东:49%,湖北:47%)。与毒力因子研究类似,对相关耐药因子的分布研究需要后期优化采样策略、扩大采样范围和样本量后进一步验证。
图3中,虚线框1代表的是菌株2014V-1011 (SAMN12648291)里的少数耐药基因,其中4个()位于该菌株一个尚未命名的质粒(CP046773.1)上。虚线框2和6中的基因分别代表新测序菌株VA28和VA24上的少数耐药基因,这些基因落在了2~3个组装片段上(contig),因此基于现有数据无法确认其在基因组上的位置是否相邻,但其中的与两个基因是Ⅰ类整合子(class 1 integron)3’端保守区域(conserved segments, 3’CS)的两个固有基因[46,47],另外虚线框1中的质粒片段上也有这两个基因,因此我们推断虚线框2和6中的少数耐药基因可能位于通过水平基因转移(horizontal gene transfer, HGT)而获得的Ⅰ类整合子片段或质粒片段上,这需要后期通过基因组完成图测序和分析进行确定。虚线框3中的基因位于菌株ZJ-T(SAMN05271497)染色体1上的相邻位置,是一个连续的大片段,我们截取这一段序列并在线BLAST比对后发现,该片段与溶藻弧菌的整合共轭元件(integrative conjugative elements, ICEs) ICEVal056-1 (accession number: KR231688.1),coverage和identity值分别为70%和99.98%,表明该菌株可能通过水平转移获得了一个携带多种耐药基因的大片段。虚线框4和5中的基因是位于菌株Vb1833 (SAMN15829766)染色体1上的两个大片段,我们分别截取了这两个片段在线BLAST后发现,比对结果中多为ICE片段或者质粒片段,这也提示菌株Vb1833 (SAMN15829766)在进化过程中,通过水平转移的方式获得了携带多种耐药基因的大片段。本研究在溶藻弧菌中发现了携带多种耐药基因的大片段,为今后溶藻弧菌所致疾病的治疗和防控提供了重要信息。

表3 部分毒力因子在不同谱系、不同国家和地区以及中国不同地点的分布统计
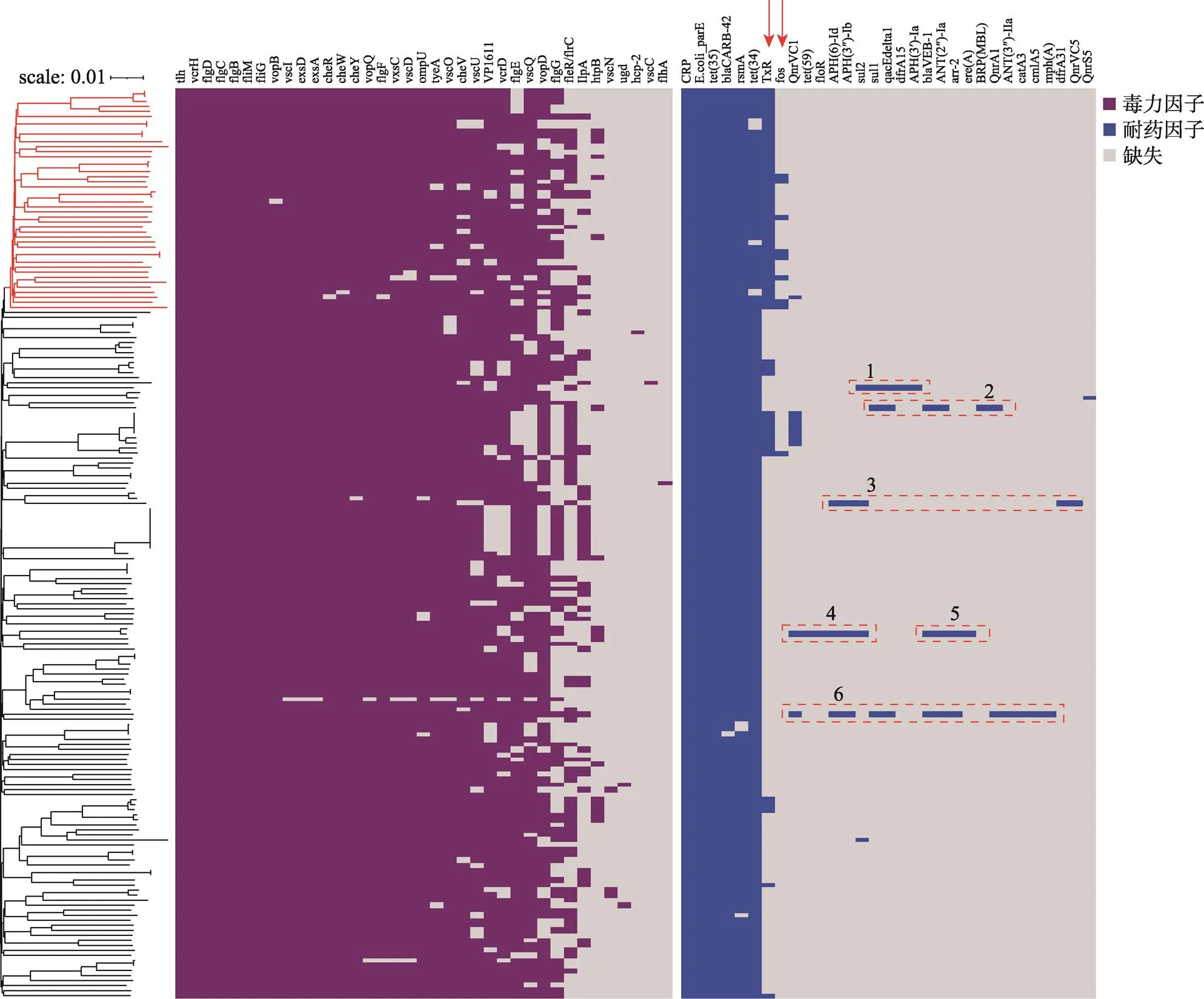
图3 181株溶藻弧菌毒力与耐药因子分布热图
紫色代表携带毒力因子,蓝色代表携带耐药因子,灰色代表缺失。虚线框代表5株菌中的少数耐药基因,红色箭头所指的两个基因为谱系2中携带率更高的两个耐药基因和。
3 讨论
过去研究发现,副溶血弧菌的种群结构与不同海域密切相关[24]。但本研究现有数据集下,溶藻弧菌不同采样地点的菌株在整个系统发育树和不同种群间未呈现地域性聚集,而是呈现出“分散分布”,即:同一采样地点的菌株(如中国、美洲等)分散于两个不同的谱系中。根据群体遗传学理论,种群分化只在种群间迁徙率很低的情况下发生(每世代中只有一个迁徙或者更少)[24],当少量迁徙者(migrant)与当地种群共存时,会逐渐与当地种群交换遗传物质,进而变得与当地种群越来越相似。本研究中种群已经形成,但两个种群(谱系1和谱系2)中还是包含了不同地域来源的菌株,可能是迁徙者到达新环境后,尚未充分与当地种群交换遗传物质,依然保留着大量“家乡”种群的遗传特征。造成该现象的原因包括:跨地域迁徙发生在近期,进化时间相对较短;或者自身遗传特征限制了遗传物质在不同种群的菌株间进行交换;或者是不同种群溶藻弧菌生活在同一地区的不同微生境下,导致事实上的生态条件隔离。将来可通过开展生态调查和实验室研究以检验上述假说。

表4 溶藻弧菌耐药基因与相关抗生素和耐药机制
括号内百分比代表整个数据集中携带此耐药基因的菌株的百分比。
本研究中,无论是从NCBI下载的公共数据,还是新测序的中国菌株,在采样地点上都存在一定的偏差。例如公共数据中,美国菌株占60%,而其他国家或大洲的菌株则相对较少,新测序的中国菌株也仅采集自中国的山东、湖北、四川和广西4个省份,其他省份数据缺失。采样偏差可能影响本研究中的某些结果。例如研究结果显示,同一谱系中(谱系1或谱系2)包含了不同大洲的菌株,提示历史上曾存在多次远距离迁徙事件,但本研究中我们仅在一个克隆群中观察到跨洋传播,可能是由于短时期迁徙事件频率低,或者是研究数据集不够完整所致。此外,种群结构研究中,发现欧洲菌株只存在于谱系1中;毒力、耐药因子分布研究中,发现某些毒力、耐药因子(、、、/、等)在不同的大洲以及中国不同的省份之间的分布存在差异。这些结果需要在将来通过完善采样策略、扩大采样范围、增加数据量等方式消除采样偏差造成的影响并进一步验证。
虽然本研究中鉴定到多种不同功能的毒力因子,但是除3株菌采集自病人外,其余均为环境菌株,因而难以将该病原的毒力因子与菌株致病性表型进行关联。此外,虽然本研究中鉴定到多种耐药相关基因,但部分鉴定结果可能为耐药基因的同源体(homology),而是否携带耐药基因同源体与菌株的临床耐药表型并不一定具有相关性,因为部分病原菌在临床上表现出对抗生素的耐受,是由染色体上的具体的基因突变导致的,包括点突变、基因片段获得缺失(Indel)等。另一方面,一个基因的表达与否还受到多种因素的调控,因此携带特定耐药基因(或同源体)的菌株是否表现出耐药表型,还要结合实验测定结果进一步研究。
综上所述,本研究利用全基因组测序技术和群体遗传学方法,对181株溶藻弧菌的遗传多样性和毒力、耐药特征进行了研究。研究结果表明溶藻弧菌是一种高频重组致病弧菌,由两个主要谱系组成,其中谱系1菌株的地域分布更为广泛。两个谱系菌株的地理分布特点和克隆群分析均表明该物种存在近期跨洋传播,但种群跨地域迁徙后尚未观察到种群融合。溶藻弧菌基因组包含多种不同功能的毒力因子,主要与溶藻弧菌自身代谢、对宿主细胞的粘附、裂解、免疫反应等功能相关,两个谱系中均未发现种群特异性毒力因子。此外,溶藻弧菌基因组既有多种核心耐药基因,也存在携带多种耐药基因的质粒或者ICE等移动遗传元件。本研究存在采样偏差以及缺乏表型实验结果等不足,但上述结果为更深入研究溶藻弧菌遗传特征和致病机制提供了基础数据,将加深对该病原的认识并促进其防控工作。此外,本研究中的研究思路和方法,也可供霍乱弧菌、河流弧菌()等其他高频重组致病菌的群体基因组学研究参考,通过揭示相关病原菌的遗传多样性、致病因子、传播模式等特征,在溯源、防控和治疗等方面提供科学支撑并发挥积极作用。
[1] Lajnef R, Snoussi M, Romalde JL, Nozha C, Hassen A. Comparative study on the antibiotic susceptibility and plasmid profiles of Vibrio alginolyticus strains isolated from four Tunisian marine biotopes., 2012, 28(12): 3345–3363.
[2] Xie JS, Bu LF, Jin S, Wang XY, Zhao QS, Zhou SM, Xu YJ. Outbreak of vibriosis caused by Vibrio harveyi and Vibrio alginolyticus in farmed seahorse Hippocampus kuda in China., 2020, 523: 735168.
[3] Reilly GD, Reilly CA, Smith EG, Baker-Austin C. Vibrio alginolyticus-associated wound infection acquired in British waters, Guernsey, July 2011., 2011, 16(42): 19994.
[4] Zhou K, Sun F, Zha DJ, Liu JY, Ma YY, Hao XK, Xu XL. Biological characteristics of a Vibrio alginolyticus strain isolated from a patient with chronic otitis externa., 2020, 158(2): 146–151.周柯, 孙菲, 查定军, 刘家云, 马越云, 郝晓柯, 徐修礼. 慢性外耳道炎患者分离溶藻弧菌的鉴定及生物学特性分析. 中国病原生物学杂志, 2020, 158(2): 146–151.
[5] Gómez-León J, Villamil L, Lemos ML, Novoa B, Figueras A. Isolation of Vibrio alginolyticus and Vibrio splendidus from aquacultured carpet shell clam (Ruditapes decussatus) larvae associated with mass mortalities., 2005, 71(1): 98–104.
[6] González-Escalona N, Blackstone GM, Depaola A. Characterization of a Vibrio alginolyticus strain, isolated from Alaskan oysters, carrying a hemolysin gene similar to the thermostable direct hemolysin-related hemolysin gene (trh) of Vibrio parahaemolyticus., 2006, 72(12): 7925–7929.
[7] Newton A, Kendall M, Vugia DJ, Henao OL, Mahon BE. Increasing rates of vibriosis in the United States, 1996-2010: review of surveillance data from 2 systems., 2012, 54 Suppl 5(05): S391–S395.
[8] Jacobs Slifka KM, Newton AE, Mahon BE. Vibrio alginolyticus infections in the USA, 1988-2012., 2017, 145(7): 1491–1499.
[9] Hu NX, Ma C, Li YJ. Research progress on molecular typing technology of Vibrio alginolyticus., 2014, 35(13): 1753–1755.胡南霞, 马聪, 李艳君. 溶藻弧菌分子分型技术的研究进展. 国际检验医学杂志, 2014, 35(13): 1753–1755.
[10] Sudheesh PS, Jie K, Xu HS. Random amplified polymorphic DNA-PCR typing of Vibrio parahaemolyticus and V. alginolyticus isolated from cultured shrimps., 2002, 207(1–2): 11–17.
[11] Ouyang JL, Zhou YC, Wu YG, Dai XL, Wang SF, Xie ZY. RAPD analysis of genetic diversity of Vibrio alginolyticus isolated from Hainan province., 2011, 35(7): 30–36.欧阳吉隆, 周永灿, 吴学贵, 戴小连, 王世锋, 谢珍玉. 海南地区溶藻弧菌遗传多样性的RAPD分析. 海洋科学, 2011, 35(7): 30–36.
[12] Ren CH, Hu CQ, Luo P, Chen C, Jiang X, Wang QB. Genotyping of Vibrio alginolyticus isolates from Daya Bay by infrequent-restriction-site PCR and pulsed-field gel electrophoresis., 2008, 22(4): 267– 271.
[13] Talon D, Cailleaux V, Thouverez M, Michel-Briand Y. Discriminatory power and usefulness of pulsed-field gel electrophoresis in epidemiological studies of Pseudomonas aeruginosa., 1996, 32(2): 135–145.
[14] Inglis TJ, Garrow SC, Adams C, Henderson M, Mayo M, Currie BJ. Acute melioidosis outbreak in Western Australia., 1999, 123(3): 437–443.
[15] Xie ZY, Hu CQ, Chen C, Zhang LP, Ren CH. Investigation of seven Vibrio virulence genes among Vibrio alginolyticus and Vibrio parahaemolyticus strains from the coastal mariculture systems in Guangdong, China., 2005, 41(2): 202–207.
[16] Hernández-Robles MF, Álvarez-Contreras AK, Juárez- García P, Natividad-Bonifacio I, Curiel-Quesada E, Vázquez-Salinas C, Quiñones-Ramírez EI. Virulence factors and antimicrobial resistance in environmental strains of Vibrio alginolyticus., 2016, 19(4): 191–198.
[17] Mechri B, Medhioub A, Medhioub MN, Aouni M. Genotypic diversity, antimicrobial resistance and screening of Vibrio cholerae molecular virulence markers in Vibrio alginolyticus strains recovered from a Tunisian Ruditapes decussatus hatchery., 2013, 62(3): 263– 272.
[18] Wei S. Detection of Vibrio alginolyticus and its virulence- related genes[Dissertation]. Jinan University, 2013.魏霜. 溶藻弧菌及其毒力相关基因的检测[学位论文]. 暨南大学, 2013.
[19] Xian YY. Analysis of the distribution of virulence-related genes and antibiotic resistance of Vibrio parahaemolyticus and Vibrio alginolyticus[Dissertation]. Jinan University, 2015.冼钰茵. 副溶血弧菌与溶藻弧菌毒力基因分布与抗生素耐药性分析[学位论文]. 暨南大学, 2015.
[20] Mechri B, Monastiri A, Medhioub A, Medhioub MN, Aouni M. Molecular characterization and phylogenetic analysis of highly pathogenic Vibrio alginolyticus strains isolated during mortality outbreaks in cultured Ruditapes decussatus juvenile., 2017, 111: 487–496.
[21] Chen HQ. Analysis of drug resistance and drug resistance genes of a pathogenic Vibrio alginolyticus from large yellow croaker., 2020, 35(1): 63–67.陈洪清. 一株大黄鱼致病性溶藻弧菌的耐药性及耐药基因分析. 渔业信息与战略, 2020, 35(1): 63–67.
[22] Limmathurotsakul D, Golding N, Dance DAB, Messina JP, Pigott DM, Moyes CL, Rolim DB, Bertherat E, Day NPJ, Peacock SJ, Hay SI. Predicted global distribution of Burkholderia pseudomallei and burden of melioidosis., 2016, 1: 15008.
[23] Chewapreecha C, Holden MTG, Vehkala M, Välimäki N, Yang ZR, Harris SR, Mather AE, Tuanyok A, De Smet B, Le Hello S, Bizet C, Mayo M, Wuthiekanun V, Limmathurotsakul D, Phetsouvanh R, Spratt BG, Corander J, Keim P, Dougan G, Dance DAB, Currie BJ, Parkhill J, Peacock SJ. Global and regional dissemination and evolution of Burkholderia pseudomallei., 2017, 2: 16263.
[24] Yang C, Pei XY, Wu YR, Yan L, Yan YF, Song YQ, Coyle NM, Martinez-Urtaza J, Quince C, Hu QH, Jiang M, Feil E, Yang DJ, Song YJ, Zhou DS, Yang RF, Falush D, Cui YJ. Recent mixing of Vibrio parahaemolyticus populations., 2019, 13(10): 2578–2588.
[25] Yang C, Zhang XL, Fan H, Li YH, Hu QH, Yang RF, Cui YJ. Genetic diversity, virulence factors and farm-to-table spread pattern of Vibrio parahaemolyticus food-associated isolates., 2019, 84: 103270.
[26] Lawson DJ, Hellenthal G, Myers S, Falush D. Inference of population structure using dense haplotype data., 2012, 8(1): e1002453.
[27] Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data., 2014, 30(15): 2114–2120.
[28] Cui YJ, Yu C, Yan YF, Li DF, Li YJ, Jombart T, Weinert LA, Wang ZY, Guo ZB, Xu LZ, Zhang YJ, Zheng HC, Qin N, Xiao X, Wu MS, Wang XY, Zhou DS, Qi ZZ, Du ZM, Wu HL, Yang XW, Cao HZ, Wang H, Wang J, Yao SS, Rakin A, Li YR, Falush D, Balloux F, Achtman M, Song YJ, Wang J, Yang RF. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis., 2013, 110(2): 577–582.
[29] Delcher AL, Salzberg SL, Phillippy AM. Using MUMmer to identify similar regions in large sequence sets., 2003, 0(1): 10.3.1–10.3.18.
[30] Seemann T. Prokka: rapid prokaryotic genome annotation., 2014, 30(14): 2068–2069.
[31] Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, Parkhill J. Roary: rapid large-scale prokaryote pan genome analysis., 2015, 31(22): 3691–3693.
[32] Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees., 2016, 44(W1): W242– W245.
[33] Liu B, Zheng DD, Jin Q, Chen LH, Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface., 2019, 47(D1): D687– D692.
[34] Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen ALV, Cheng AA, Liu SH, Min SY, Miroshnichenko A, Tran HK, Werfalli RE, Nasir JA, Oloni M, Speicher DJ, Florescu A, Singh B, Faltyn M, Hernandez-Koutoucheva A, Sharma AN, Bordeleau E, Pawlowski AC, Zubyk HL, Dooley D, Griffiths E, Maguire F, Winsor GL, Beiko RG, Brinkman FSL, Hsiao WWL, Domselaar GV, Mcarthur AG. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database., 2020, 48(D1): D517–D525.
[35] Bortolaia V, Kaas RS, Ruppe E, Roberts MC, Schwarz S, Cattoir V, Philippon A, Allesoe RL, Rebelo AR, Florensa AF, Fagelhauer L, Chakraborty T, Neumann B, Werner G, Bender JK, Stingl K, Nguyen M, Coppens J, Xavier BB, Malhotra-Kumar S, Westh H, Pinholt M, Anjum MF, Duggett NA, Kempf I, Nykäsenoja S, Olkkola S, Wieczorek K, Amaro A, Clemente L, Mossong J, Losch S, Ragimbeau C, Lund O, Aarestrup FM. ResFinder 4.0 for predictions of phenotypes from genotypes., 2020, 75(12): 3491–3500.
[36] Wang HQ, Yang C, Sun Z, Zheng W, Zhang W, Yu H, Wu YR, Didelot X, Yang RF, Pan JC, Cui YJ. Genomic epidemiology of Vibrio cholerae reveals the regional and global spread of two epidemic non-toxigenic lineages., 2020, 14(2): e0008046.
[37] Cui YJ, Yang C, Qiu HL, Wang H, Yang RF, Falush D. The landscape of coadaptation in Vibrio parahaemolyticus., 2020, 9: e54136.
[38] Zhao Z, Liu JX, Deng YQ, Huang W, Ren CH, Call DR, Hu CQ. The Vibrio alginolyticus T3SS effectors, Val1686 and Val1680, induce cell rounding, apoptosis and lysis of fish epithelial cells., 2018, 9(1): 318–330.
[39] Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Iijima Y, Najima M, Nakano M, Yamashita A, Kubota Y, Kimura S, Yasunaga T, Honda T, Shinagawa H, Hattori M, Iida T. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae., 2003, 361(9359): 743–749.
[40] Zhou XH, Konkel ME, Call DR. Type III secretion system 1 of Vibrio parahaemolyticus induces oncosis in both epithelial and monocytic cell lines., 2009, 155(Pt 3): 837–851.
[41] Burdette DL, Seemann J, Orth K. Vibrio VopQ induces PI3-kinase-independent autophagy and antagonizes phagocytosis., 2009, 73(4): 639–649.
[42] Jia AR, Woo NYS, Zhang XH. Expression, purification, and characterization of thermolabile hemolysin (TLH) from Vibrio alginolyticus., 2010, 90(2): 121–127.
[43] Wong SK, Zhang XH, Woo NYS. Vibrio alginolyticus thermolabile hemolysin (TLH) induces apoptosis, membrane vesiculation and necrosis in sea bream erythrocytes., 2012, 330–333: 29–36.
[44] Cai SH, Yaol SY, Lu YS, Wu ZH, Jian JC, Wang B. Immune response in Lutjanus erythropterus induced by the major outer membrane protein (OmpU) of Vibrio alginolyticus., 2010, 90(1): 63–68.
[45] Lv TT, Dai F, Zhuang QT, Zhao XL, Shao YN, Guo M, Lv ZM, Li CH, Zhang WW. Outer membrane protein OmpU is related to iron balance in Vibrio alginolyticus., 2020, 230: 126350.
[46] Cambray G, Guerout AM, Mazel D. Integrons., 2010, 44: 141–166.
[47] Rajpara N, Nair M, Bhardwaj AK. A highly promiscuous integron, plasmids, extended spectrum beta lactamases and efflux pumps as factors governing multidrug resistance in a highly drug resistant Vibrio fluvialis isolate BD146 from Kolkata, India., 2018, 58(1): 60–67.
Population genomics study of
Hongyuan Zheng1, Lin Yan2, Chao Yang1,3, Yarong Wu1, Jingliang Qin1, Tongyu Hao1, Dajin Yang2, Yunchang Guo2, Xiaoyan Pei2, Tongyan Zhao1, Yujun Cui1
is a Gram-negative bacillus that causes vibriosis to human and aquatic products, including fish, shrimp and shellfish. It poses a threat to public health and causes enormous economic losses to the aquaculture industry. However, research on genetic diversity and pathogenicity-related genetic elements based on whole genome is still lacking.In this study, sixty-eight strains ofwere collected from four provinces of China and the whole genome sequences were obtained. Combined with 113 publicly available genome sequences downloaded from NCBI, we inferred the population structure ofby using fineSTRUCTURE software, and identified the virulence and antibiotic resistance factors using the VFDB, CARD and ResFinder database. The results indicated thatincluded two main lineages, named Lineage 1 and Lineage 2. Both lineages distributed in America and Asia, but all the European genomes were classified into Lineage 1. A single cross-ocean transmission event was inferred from one of the 12 identified clonal groups in our dataset.genome contains a variety of virulence factors, such as,, andetc. The distribution of virulence factors revealed no lineage-specificity, but some of which revealed differences in their geographical distribution. A lower frequency of,,,and a higher frequency ofwere observed in genomes of Europe than other continents. In China, a lower frequency of,and nowere observed in genomes from Guangxi province. Among the identified antibiotic resistance genes,andare significantly enriched in Lineage 2. In addition,is more common in genomes from Asia, compared with the American and European genomes. But in China, the frequency ofin Sichuan genomes is much lower than in other provinces. We also found that large fragments of plasmids or ICEs that carried multiple drug resistance genes were present in fivegenomes (VA24, VA28, 2014V-1011, ZJ-T and Vb1833). Based on population genomics analysis, our study delineated the population structure, distribution of virulence and antibiotic resistance related factors of, which lays a foundation for future study of genetic characters and pathogenesis mechanism of this pathogen and will improve the works on monitoring, prevention and control of this pathogen.
;whole genome sequencing; population structure; virulence factor; antimicrobial resistance gene
2021-02-09;
2021-03-16
国家重点研发计划(编号:2018YFC1603902,2017YFC1601500)和中国博士后科学基金项目(编号:2020M672836)资助[Supported by the National Key R&D Program (Nos. 2018YFC1603902, 2017YFC1601500) of China, and China Postdoctoral Science Foundation (No. 2020M672836)]
郑宏源,在读硕士研究生,专业方向:细菌基因组流行病学。E-mail: zhenghongyuan.chn@qq.com
崔玉军,博士,研究员,研究方向:微生物进化与基因组流行病学。E-mail: cuiyujun.new@gmail.com
10.16288/j.yczz.21-061
2021/4/1 15:08:36
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20210401.1103.004.html
(责任编委: 高海春)
猜你喜欢
当代水产(2022年8期)2022-09-20
艺术品鉴(2022年16期)2022-07-09
食品安全导刊(2021年20期)2021-08-30
河池学院学报(2021年1期)2021-07-10
景德镇陶瓷(2021年1期)2021-03-24
当代水产(2019年5期)2019-07-25
东方考古(2017年0期)2017-07-11
西南农业学报(2017年5期)2017-06-23
环境污染与防治(2016年9期)2016-03-13
微生物学杂志(2015年6期)2015-12-27
