
含磷多孔有机聚合物的合成及其在多相催化中的应用
2021-04-20 10:30吴淼江孙鹏李福伟
化工进展 2021年4期
吴淼江,孙鹏,李福伟
(1 中国科学院兰州化学物理研究所羰基合成与选择氧化国家重点实验室,甘肃兰州730000;2 中国科学院大学,北京100049)
膦配体(本文指三价有机膦类、亚磷酸酯类或亚磷酰胺类配体)既是σ电子给体也是π电子受体(图1)[1],作为一种“软碱”与后过渡金属钌[2]、锇[3]、钴[4]、铑[5]、铱[6]、镍[7]、钯[8]、铂[9]等有着很好的配位能力。金属-膦配合物作为均相催化剂在碳氢键、饱和碳碳键、不饱和碳碳键、碳氮键、碳氧键、碳卤键等的活化或构建中有着广泛的应用,而且在反应中常常能表现出很高的催化活性和选择性。但它们溶解于反应体系中难以分离回收,大大限制了其在大规模生产中的应用。

图1 有机膦配体的电子性质
为了方便金属-膦配合物催化剂的回收和重复利用,科研人员尝试将膦配体锚定在有机高分子载体或无机载体上,制备锚定的金属-膦配体催化剂(图2)。其中,用可溶性聚合物锚定膦配体是具有代表性的方法之一。可溶性聚合物能溶于反应体系,与均相催化剂极为接近,官能团化的膦配体很容易通过接枝或共聚的方式引入,因此20 世纪末报道了大量的可溶性聚合物基金属-膦配体催化剂。常见的用于锚定膦配体的可溶性聚合物有线性聚苯乙烯[图2(a)][10]、线性聚乙二醇[图2(b)][10]、肽链聚合物[图2(c)][10]以及支链聚合物[图2(d)][11]等。但可溶性聚合物基催化剂需要通过添加沉淀剂、微滤或超滤等手段来实现催化剂的回收,且小分子聚合物链有可能残留于产物中产生污染,因此逐渐被研究人员所舍弃。随着无机-有机杂化材料的兴起,科研人员将官能团化的膦配体通过吸附或者化学键合的方式与无机材料结合制备含磷多相催化剂。其中,具有代表性的无机材料有高比表面积的有序介孔硅材料[图2(e)、(f)][12]和具有磁性的Fe3O4纳米颗粒[图2(g)][13]。但是无机材料表面活性位点单一,化学修饰潜力十分有限,大大限制了基于膦配体-无机载体催化剂的开发。
多孔有机聚合物(porous organic polymers,POPs)是由有机小分子通过共价键联结而成的孔基材料。相较于可溶性聚合物,多孔有机聚合物具有比表面积大、孔隙发达和易于从溶剂体系中分离的优势;相较于有机-无机杂化材料,它具有分子骨架稳定、可调变性和可修饰性强的优势。因此,多孔有机聚合物在分离、多相催化、污染物捕集、气体储存等方面有着广泛的应用前景[14]。

图2 传统方式锚定膦配体示例
多孔有机聚合物中微孔、介孔和大孔往往同时存在,这种多级孔的特征特别适合于用作多相催化剂载体[15],并能结合均相催化体系的某些优势[16]。由于多孔有机聚合物的结构易于调变,其在仿生催化材料方面具有开发潜力[17]。迄今为止,虽已有一些关于多孔有机聚合物的制备及其应用的综述发表[14,18-19],但还没有概述和总结含磷多孔有机聚合物及其在多相催化中应用的综述发表(本文所述聚合物单体包含三价有机膦类、亚磷酸酯类、磷杂苯类、氯化磷类和季盐类,因此用意含磷元素的“磷”命名聚合物,即“含磷多孔有机聚合物”)。事实上,作为多相催化剂制备新平台,近十年来含磷多孔有机聚合物载体的发展十分迅速,甚至在工业化生产中已有应用。中国科学院大连化学物理研究所丁云杰和严丽团队[20]基于含磷多孔聚合物基催化技术,实现了乙烯氢甲酰化制备丙醛/正丙醇的多相催化,推动5 万吨/年的制备丙醛/正丙醇工业装置成功投产。研究人员在膦配体单体设计、聚合物合成方法等方面做了大量的工作,含磷多孔有机聚合物已成为一个有活力且极具发展潜力的领域。因此,很有必要对近十年来含磷多孔有机聚合物基催化剂的制备和应用进行系统性整理。结合本文作者课题组在卡宾聚合物基催化剂方面的理解[21-25],本文对含磷多孔聚合物基催化剂设计、合成以及应用过程中发现的一些规律进行归纳和阐述。
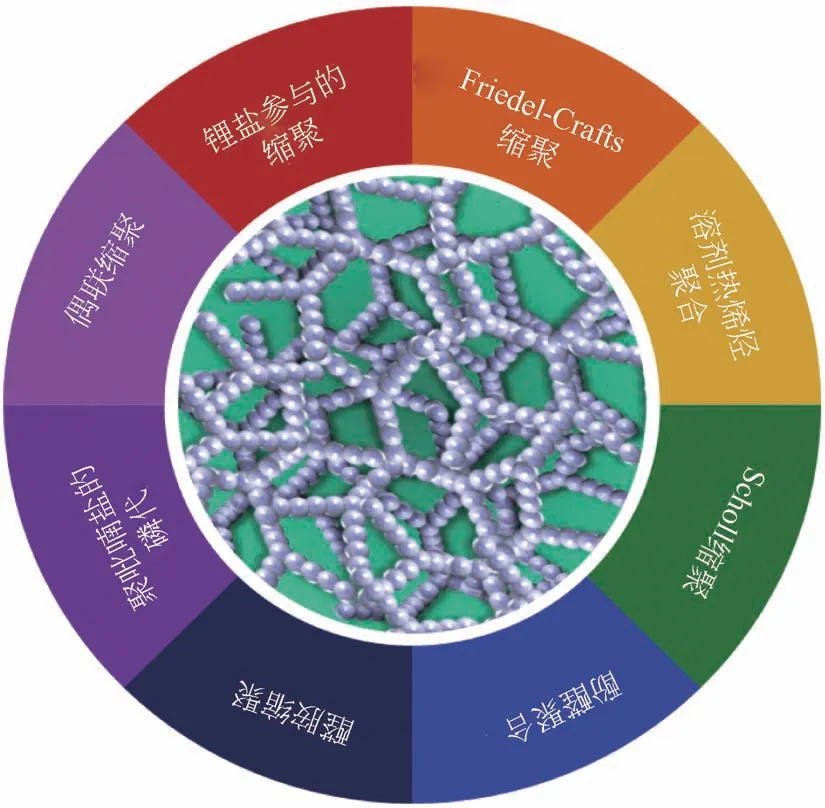
图3 含磷多孔有机聚合物合成方法
目前为止,文献报道的含磷多孔有机聚合物制备方法有偶联缩聚、锂盐参与的缩聚、Friedel-Crafts 缩聚、溶剂热烯烃聚合、Scholl 缩聚、酚醛聚合、醛胺缩聚、聚吡喃盐的磷代以及多段式聚合等(图3)。多元的聚合方法意味着聚合单体结构的多样化,使得聚合单体的设计和开发十分灵活多变。而合成方法的多样性以及合成单体的多变性又使得含磷多孔有机聚合物具有巨大的调变空间,可以根据各种催化反应体系的特点有针对性、有目的性地设计和调变催化剂结构。这使得制备高活性、高选择性的催化剂成为可能。可调变性和可修饰性强是含磷多孔有机聚合物催化剂的优势,同时也是此类催化剂发展面临的难点和挑战:如何在如此多变的体系中找到适合反应体系的催化剂?围绕这一核心问题,本文对近十年来含磷多孔有机聚合物载体的制备方法、聚合单体进行了总结,旨在为含磷多孔有机聚合催化剂的设计合成提供些许指导和帮助,进一步加快此类催化剂的发展及工业应用进程。
多孔有机聚合物的分类和命名十分繁杂,如微孔有机聚合物(microporous organic polymers,MOPs)、自具微孔聚合物(polymers of intrinsic microporosity, PIMs) 、 超 交 联 聚 合 物(hypercrosslinked polymers,HCPs)、共轭微孔聚合物(conjugated microporous polymers,CMPs)、共价有机框架(covalent organic frameworks,COFs)、编织芳基网络聚合物(knitting aryl network polymers,KAPs)、多孔离子聚合物(porous Ionic Polymers,PIPs)等。在某些情况下,各种方式命名的多孔聚合物之间不能明确地界定。为使叙述更加清晰明了,本文以制备方法进行分类来对含磷多孔有机聚合物进行综述。
1 偶联缩聚
偶联缩聚是具有多反应位点的单体间通过偶联形成碳碳键交联在一起,是制备多孔聚合物的常用方法,这类反应包括Heck 偶联、Sonogashira-Hagihara 偶联、Suzuki-Miyaura 偶联和Yamamoto 偶联等(图4)[26]。基于此,一系列具有多反应位点的膦配体单体和共聚单体被合成,并用于含磷多孔有机聚合物的制备。
1.1 Heck偶联缩聚
Heck 偶联缩聚是利用多卤代芳烃单体和多乙烯基芳烃单体间的交叉偶联,进而高度交联成聚合物的方法。2015年,张帆团队[27]分别用膦配体单体a1、a2和共聚单体b1,通过Heck偶联缩合“一锅”合成了含磷多孔有机聚合物基钯纳米颗粒催化剂。该催化剂在Suzuki-Miyaura 偶联反应中表现出很好的活性,分别基于a1 和a2 的聚合物催化剂在Suzuki-Miyaura 偶联反应中表现出相似的催化活性,体系中磷的价态对钯物种催化活性的影响较小。值得注意的是,该文还指出在一定范围内偶联缩聚钯催化剂用量增加能促进缩聚反应的进行,从而使得聚合物孔径减小、比表面积增大。但钯催化剂用量过高,又可能因钯颗粒占据或堵塞孔结构导致比表面积减少。2017年,詹庄平团队[28]使用乙烯基化的膦配体单体a3和溴代单体b2,经Heck偶联缩合、碳碳双键还原两步成功合成了含磷多孔聚合物。由于骨架结构中碳碳双键被还原成了更具柔性的单键,该聚合物孔径较上述张帆团队合成的聚合物具有更大的孔径。在多相催化中,较大的孔道有利于底物在催化剂中的扩散,从而更快地接触到活性位点。该聚合物负载钯纳米颗粒后,在硝基芳烃、α,β-不饱和烯烃等化合物的加氢反应中表现出高活性、好的底物适用性和重复使用性。

图4 偶联反应
1.2 Sonogashira-Hagihara偶联缩聚
为了防止炔烃单体间的自身偶联,Sonogashira-Hagihara偶联缩聚必须在严格无氧的条件下进行。2015 年,李灿团队[29]使用(R)-4,4-DibromoBINAPO 单体(a4),分别同b3、b4、b5 和b6 通过Sonogashira-Hagihara 偶联缩聚、三氯硅烷脱氧两步成功合成了一系列含BINAP 的聚合物载体。实验结果表明,共聚单体对催化剂性能影响明显:基于共聚单体b6 的聚合物与[RuCl2(benzene)]2配位后在β-酮酯的不对称加氢反应中表现出很好的活性和手性选择性,转化率和对映体过量值(enantiomeric excess,ee) 均能达到99%;其与[Ir(COD)Cl2]配位后,在喹哪啶的不对称加氢中表现出比均相催化体系(BINAP/[Ir(COD)Cl]2)更高的催化活性和相当的选择性。该项工作说明通过膦配体单体调控聚合物基催化剂活性和选择性是可行的。
1.3 Suzuki-Miyaura偶联缩聚
2017 年,李涛团队[30]用a5 和b7 通过Suzuki-Miyaura 偶联缩聚“一锅”合成了聚合物基钯纳米颗粒催化剂,在水和乙醇的混合溶剂中对Suzuki-Miyaura 偶联反应表现出很好的催化活性和良好的底物适用性。该催化剂重复使用5次后,XPS谱图中零价钯比例显著上升,说明钯发生了团聚。2019年,Bojdys 团队[31]通过Suzuki-Miyaura 偶联缩聚,使用甲氧基稳定的λ5磷杂苯类化合物a6 和b7 成功合成了π 共轭的λ5磷杂苯共价网络聚合物(covalent phosphinine framework,CPFs)。以三乙醇胺为牺牲剂,在波长380~780nm的可见光条件下,该聚合物在钯(合成过程中残留)的助催化下光解析氢效率速率为33.3mmol/(h·g)。要拓展磷杂苯类聚合在多相催化中的应用,必须要合成比λ5磷杂苯具有更强配位能力的λ3磷杂苯聚合物。但实际上,甲氧基稳定的λ5磷杂苯单体结构也很容易被酸、碱破坏,而λ3磷杂苯是更不稳定的单体分子。因此,从λ3磷杂苯小分子单体出发合成含磷杂苯的聚合物是很困难的。
1.4 Yamamoto偶联缩聚
2012 年,张所波团队[32]使用季盐单体a8 通过Yamamoto 自偶联缩聚制得了季盐型多孔有机聚合物。该聚合物直接使用能催化环氧化合物与二氧化碳生成环碳酸酯的反应,负载钯纳米颗粒后能用于催化Suzuki-Miyaura 偶联反应。次年,他们[33]又用a5、a7分别合成了两种比表面积超过1200m2/g的多孔聚合物,该聚合物500℃条件下还能保持稳定。通过浓盐酸处理和溶剂洗涤的方式除去镍金属缩聚催化剂,再引入钯物种制成聚合物基钯纳米催化剂,能催化Suzuki-Miyaura 偶联反应的进行。与前文张帆团队[27]的实验结果不同,磷物种价态对张所波团队制备的催化剂活性有着明显影响:基于三价膦单体(a5)的聚合物催化剂活性明显高于基于五价的氧磷单体(a7)的聚合物催化剂,说明三价膦配体促进了钯物种在该反应体系中的催化活性。
近十年来通过偶联缩聚制备的含磷多孔有机聚合物,其单体组分、物理性质(比表面积、孔体积和孔径)、负载金属类型以及相应催化剂的应用参见图5和表1。

图5 偶联缩聚单体

表1 偶联缩聚含磷聚合物单体组成、孔结构参数及其在多相催化中的应用
前文例举的一些文献结果表明,含磷单体和共聚单体均对聚合物物理性质(比表面积、孔体积、孔径分布等)有影响,且与聚合物基催化剂催化性能密切相关。缩聚催化剂的量也能影响聚合物物理性质。催化剂量越大越有利于缩聚,一定范围内随缩聚催化剂量增加,合成的聚合物孔径变小、比表面积增大[27]。由表1可知,通过偶联缩聚合成的含磷多孔有机聚合物孔径分布普遍较窄,主要由微孔和孔径偏小的介孔组成[参照国际纯粹与应用化学联合会(IUPAC)对多孔材料的分类:微孔<2nm、介孔2~50nm、大孔>50nm],说明偶联缩聚制得的聚合物高度交联。值得注意的是,不同偶联缩聚方式合成聚合物的性质也有所差别。Suzuki-Miyaura和Yamamoto偶联缩聚合成的含磷多孔有机聚合物,单体之间由苯环间的碳碳键直接相连,具有很强的刚性。而Heck、Sonogashira-Hagihara 偶联缩聚合成的多孔聚合物中的碳碳双建和三键还原后变成柔性的烷基链段,相比Suzuki-Miyaura 和Yamamoto偶联缩聚理论上能得到孔径更大、溶胀性更好的聚合 物。 此 外, Heck、 Sonogashira-Hagihara 和Suzuki-Miyaura 偶联缩聚需要钯催化剂的参与,Yamamoto 偶联缩聚需要镍催化剂的参与,而钯和镍都能与膦配体配位并且会在催化过程中团聚,这势必给负载其他金属带来麻烦。从表1 可以看出,通过偶联缩聚制备的聚合物基催化剂负载金属大部分是钯,单一的金属负载一定程度上限制了其在多相催化领域的应用。如果在钯或镍的基础上再引入其他金属物种应用在某些特定的领域(如需要多活性位点的串联催化),不仅能拓宽偶联缩聚聚合物基催化剂的应用,还能省去去除缩聚催化剂的步骤。
2 锂盐参与的缩聚
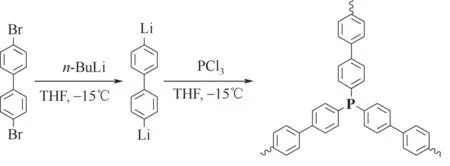
图6 锂盐参与的缩聚
2008年,Kaskel课题组[34]首次通过二取代联苯锂盐与硅酸乙酯反应制得多孔聚合物。2012 年,他们[35]将该方法用于含磷多孔有机聚合物的合成。如图6,二溴代联苯(a9)与正丁基锂反应生成二锂代联苯,再与三氯化磷(b8)偶联缩聚得到含磷聚合物。该聚合物负载RhCl(PPh3)3后用于环己酮与异丙醇的氢转移反应,二次使用时活性明显下降,催化剂稳定性差。2013 年,Bruijnincx 团队[36]同样用单体a9 和b8 合成了含磷聚合物,并用苯基锂进一步除去聚合物中残留的氯元素。该文指出钯前体对其在聚合物中的分散形式影响明显。聚合物负载零价的Pd(dba)2后从电镜照片观察到钯以纳米颗粒的形式分散在聚合物中,负载二价的Pd(acac)2则没有发现以纳米颗粒存在的钯物种,不过以Pd(acac)2为前体制备的催化剂在使用过程中或Pd(acac)2过量的情况下还是会有钯纳米颗粒的生成。该作者还研究了钯与磷元素的比例对钯配位方式的影响。磷/钯比小于2时,13P核磁谱图中未配位的磷与配位磷的信号峰共存;磷/钯比接近2 时,13P 核磁谱图中未配位磷的信号峰基本消失。虽然不能确定钯与磷是否为二齿配位,但却证明了该聚合物中几乎所有磷位点都能与钯配位,说明该法合成的含磷聚合物中磷位点极少被聚合物骨架包埋。2016 年,Palkovits 团队[37]通过锂盐缩聚的方法制备了用于甲酸分解制氢的聚合物基钌催化剂。该文指出基于双膦单体a10的钌基聚合物催化剂催化活性和选择性均优于基于a9和a11的催化剂,重复使用7次后只有少许Ru 纳米颗粒出现,具有良好的稳定性。对比以上3个团队制备的聚合物比表面积(表2),后两者采用了苯基锂后处理的缩聚物比表面明显要低一些,说明苯基取代未参与反应的氯原子的同时会占据相当一部分的孔体积。

表2 锂盐缩聚含磷聚合物单体组成、孔结构参数及其在多相催化中的应用
2019 年,Yoon 团队[38]使用四反应位点的单体b9 同双膦单体a10 制得了含磷聚合物。相较于b8,b9是一个具有更多反应位点的立体型的交联单体,制得的聚合物即使经苯基锂处理,其比表面积和孔体积依然能达到469m2/g和0.475cm3/g。该聚合物引入RuCl3后制得的催化剂在二甲胺的氢甲酰化反应(二氧化碳为羰源)中表现出很好的活性和稳定性。鉴于RuCl2(dppe)2早就被证明是该反应的非常有效的均相催化剂[39-40],而Yoon团队所制备的聚合物中包含类1,2-双(二苯基膦)乙烷(dppe)的结构。这说明均相催化理论能对含磷聚合物基催化剂单体的设计和选择提供方向和指导。
本部分提到的单体、聚合物物理性质以及各聚合物基催化剂的应用汇总见图7和表2。
图7 锂盐缩聚单体
锂盐与氯化磷的缩聚反应比较容易进行且不可逆,理论上会使得单体之间连接的无序度增加。与偶联缩聚聚合物相比,该类聚合物孔道更大且分布不均匀。表2中聚合物孔径分布就很好地验证了这一点,其氮气吸附等温线呈典型的多级孔分布(详见对应参考文献)。此外,正丁基锂、苯基锂和氯化磷对水分极其敏感,反应活性高,使得此类缩聚反应具有一定危险性。
3 Friedel-Crafts缩聚
Friedel-Crafts缩聚是芳香族化合物在路易斯酸的催化下,通过偶联剂相互联结形成超交联网状无规聚合物的方法。自2012 年谭必恩和李涛团队[41]首次将该方法应用于合成含磷多孔有机聚合物合成以来,科研人员一直试图拓展该方法在多相催化领域的应用。Friedel-Crafts缩聚能合成含大孔、介孔和微孔的多级孔含磷多孔聚合物载体,也能通过选择合适的缩聚单体和偶联剂制备微孔或介孔含磷多孔聚合物载体。
3.1 膦配体间的Friedel-Crafts缩聚

图8 单体a12、b10和c1间的Friedel-Crafts缩聚
2012 年,谭必恩团队用单体a12、b10 和偶联剂c1 在氯化铁的催化下首次成功合成了比表面积高达1036m2/g 的多级孔KAPs(Ph-PPh3)[图8(a)],膦配体均匀分散于KAPs(Ph-PPh3)体相中。该聚合物负载PdCl2后用于卤代芳烃[41]或氯化苄[42]与苯硼酸类化合物的Suzuki-Miyaura 交叉偶联反应表现出很高的活性。多级孔载体中大孔和介孔有利于传质,微孔则有利于金属的均匀分布和稳定。为了阐述微孔对催化剂稳定性能的作用,他们还使用另一种方式制备聚合物基钯催化剂作为比较。如图8(b),该作者先用b10和c1缩聚合成KAPs(Ph),然后再加入含磷单体a12 进行第2 步缩聚合成KAPs(Ph)-PPh3。该法制备的聚合物中膦配体更多的是分散于聚合物表面,两种KAPs 负载PdCl2后分别记为KAPs(Ph-PPh3) -Pd 和KAPs(Ph) -PPh3-Pd, 用 作Suzuki-Miyaura 交叉偶联催化剂。实验表明,后者钯流失严重,使用1次就有钯黑生成,第2次使用时活性明显降低;而前者首次使用钯流失率仅约1%,重复使用5次后催化活性无明显下降,催化剂电镜照片与新鲜催化剂没有明显变化。这说明微孔能有效防止钯的流失和聚集,从而提高其稳定性。随后,他 们 又 将RuCl3·H2O 负 载 于KAPs 用 于NH4OAc、DMF 与苯乙酮的环化反应以及重氮二羰基化合物与烯烃的环加成反应[43]。该催化剂同样表现出很好的稳定性,重复使用7 次催化剂中钌无明显流失。
2014 年,丁云杰团队[44]改变a12、b10 和c1 比例制得了用于高碳烯烃氢甲酰化的铑纳米颗粒催化剂。该研究指出,基于KAPs 的铑催化剂较基于二氧化硅的催化剂具有更大比表面积和孔体积,铑纳米颗粒在KAPs 中的分布更加均匀,具有更高的催化活性。但是该催化剂化学选择性和区域选择性差,有大量烷烃和异构烯烃生成,直链产物和支链产物之比也处在0.5~1的范围内。
随后,双膦配体a13[45]、含S/N杂原子的a14[46]、a15[47]的膦配体以及一些含杂原子的共聚单体[46,48]也被应用到含磷KAPs的制备中。景晓飞团队[47]发现,改变膦配体单体能影响聚合物负载的金纳米颗粒粒径。基于含吡啶基膦配体(a15)的KAPs中生长的金纳米颗粒粒径在1.9nm 左右,而基于三苯基膦(a12)的粒径分布则在更大2.8nm。无独有偶,黄延强团队[48]也在制备KAPs 负载的银纳米颗粒催化剂时发现,基于三苯基膦(a12)的KAPs中生长的银纳米颗粒粒径均匀分布在4.1nm左右,而无膦配体单体时银纳米颗粒粒径在9.8nm左右,且粒径分布不均匀。由此可见,制备基于KAPs 的金属纳米颗粒催化剂时,能通过改变缩聚单体对纳米金属颗粒粒径的大小和分布进行调控,从而调控其催化性能。
3.2 金属-膦配合物间的Friedel-Crafts缩聚
2017年,赖志平团队[49]将Friedel-Crafts缩聚应用到了金属-膦配合物的缩聚中来,用钯配合物a16 一步合成了单位点的PPPd 催化剂(图9)。该催化剂在偶联反应(Suzuki-Miyaura 交叉偶联、卤代芳烃还原自偶联、芳烃硼酸氧化自偶联)和C—H 键官能团化(烷氧基化、卤化、烯基化)反应中表现出良好的催化活性和稳定性。2018 年,该团队[50]又使用钌配合物a17 一步合成用于甲酸制氢的钌催化剂,并通过EXAFS 分析证明了该催化剂钌主要以单原子的形式存在。该催化剂在DMSO、H2O/DMSO 溶剂体系下循环使用50 次甲酸转化率未见明显下降,表现出很好的稳定性。值得指出的是,该团队此前已经证明配合物a17是甲酸制氢的很好的均相催化剂[51]。这再次体现出均相对聚合物基催化剂的指导作用。2019 年,Iglesias 团队[52]将该方法应用到更多的金属配合物,如a18、a19 和a20,制备了一系列含钌、金的催化剂。直接用金属配合物通过Friedel-Crafts缩聚合能合成高金属/磷比例的金属催化剂,相较金属后负载的方式其操作也更加简便,控制得当还能制备单原子催化剂。

图9 一步法合成单位点钯催化剂PPPd
一方面,Friedel-Crafts 缩聚无需使用官能基化的单体而选择范围广,能通过调控单体组分(膦配体单体、非膦共聚单体和偶联剂及其配比)控制含磷聚合物比表面积和孔径分布等性质;另一方面,Friedel-Crafts 缩聚合成步骤简单,既能先合成含磷聚合物再通过后负载制备金属催化剂,也能直接用金属配合物一步合成,在含磷多孔有机聚合物基催化剂的制备上具有一定的普适性。但是,Friedel-Crafts 缩聚需要使用FeCl3或AlCl3做催化剂,如果除不净可能对反应带来不利影响。此外,通过该法调控膦配体单体改变金属空间位阻进而控制催化剂区域选择性的报道还没有,还需更系统的研究。
本部分提到的单体、聚合物物理性质以及各聚合物基催化剂的应用汇总见图10和表3。

图10 Friedel-Crafts缩聚单体

表3 Friedel-Crafts缩聚含磷聚合物单体组成、孔结构参数及其在多相催化中的应用
4 溶剂热烯烃聚合
早期,通过烯烃自由基聚合制备含磷聚合物载体以可溶的线性聚苯乙烯为代表[10]。随后,科研人员又利用多烯烃反应位点的膦配体单体通过悬浮聚合[53]或乳液聚合[54]等途径合成一些交联的孔基含磷聚合物载体。一般说来,这类聚合物的比表面积较小,孔结构不发达。2009年,肖丰收团队[55]首次报道了溶剂热聚合的方法。溶剂热烯烃聚合,选用适当的溶剂能合成具有发达孔道结构和高比表面积的聚合物。多孔性的含磷聚合物十分适合用作催化剂载体,因此该法在含磷多孔有机聚合物基的催化剂制备中被广泛应用。迄今为止,被报道的用于溶剂热烯烃聚合的烯基化的含磷单体已经有数十种,包括烯基化的单膦配体、双膦配体和季盐单体。本文总结了通过溶剂热方式制备的多孔聚合物,其单体组分、物理性质(比表面积、孔体积和孔径)、负载金属类型以及相应催化剂的应用,在相应部分的图表列出。
4.1 单膦配体聚合物
铑与膦配体(三价膦配体、亚磷酸酯配体和亚磷酰胺配体)的配合物是一类活性很高的烯烃氢甲酰化催化剂,例如已经工业应用的HRh(CO)(PPh3)2催化剂,它能在低一氧化碳压力条件下催化氢甲酰化反应高效进行[56]。2014—2015年,肖丰收团队和丁云杰团队[57-58]制备了一系列用于氢甲酰化反应的含磷聚合物基铑催化剂,并对聚合物基催化剂体系进行了系统的研究。该作者用膦配体单体3V-PPh3(a21),通过溶剂热聚合成功制得比表面积和孔体积分别高达1086m2/g 和1.70cm3/g 的含磷聚合物(POL-PPh3),负载Rh(CO)2(acac)和HRh(CO)(PPh3)3后分别获得了单位点铑的氢甲酰化催化剂Rh(CO)2(acac)/POL-PPh3和RhH(CO)(PPh3)3/POL-PPh3。两种聚合物基催化剂表现出与均相催化剂[HRh(CO)(PPh3)3]类似的活性和选择性,且在釜式反应器和固定床反应器中均能使用。其中,Rh(CO)2(acac)/POL-PPh3于固定床反应器催化乙烯的氢甲酰化 反 应, TOF (turnover frequencies) 值 能 达10534h-1[57]。根据Rh(CO)2(acac)/POL-PPh3在混合气体(C2H4∶CO∶H2=1∶1∶1)中的原位红外分析结果,再结合其与HRh(CO)(PPh3)3表现出类似活性和选择的实验结果,提出了铑在聚合物中与三个磷原子配位的模型(图11)。随后,丁云杰团队[59]用原位加入和后负载铑配合物两种方式制备催化剂,发现原位引入方式可能使铑位点被聚合物覆盖而活性较低。接着,肖丰收团队[60]探究了聚合物中磷的浓度以及铑/磷比例对催化活性的影响。实验结果表明高磷浓度的聚合物载体有利于催化剂活性的提高,而铑/磷比例过高或过低都会给催化剂带来不利影响。

图11 Rh/POL-PPh3中铑、磷之间的三角锥和平面配位模型
2015—2017年,丁云杰团队又将PdCl2(PhCN)2[61]、RuCl3[62]和Rh2(CO)4Cl2[63]分别负载于POL-PPh3上应用于催化Suzuki-Miyaura 偶联反应、肉桂醛加氢反应和甲醇羰化反应。2018 年,雷以柱团队[64]将Pd(OAc)2负载于POL-PPh3上用于催化苯硼酸化合物或醇类化合物与卤代芳烃的羰化偶联。
含磷聚合物载体通过单体设计和共聚的方法赋予其特殊性质,使得聚合物基催化剂具有很好的可调变性。丁云杰团队[65]通过用a21 与b18~b20 共聚合成一系列用于加速环氧化合物与二氧化碳生成环碳酸酯的催化剂。在甲酯基化反应中,吡啶基取代的膦配体能加速醇的脱质子从而加速酯的生成[66]。基于此,丁云杰团队[67]合成了基于单体a22的含氮、磷的多孔聚合物,负载Pd(OAc)2后用于炔烃的甲酯基化反应。之后他们使用a21 或a22 与b16 共聚引入苯磺酸基团获得了多功能甲酯基化反应催化剂[68]和吗啉氢甲酰化反应(二氧化碳为羰源)催化剂[69]。近来,他们又通过筛选大量基于不同烯基化膦配体单体(a23~a33)的含磷多孔聚合物,开发出了一种用于末端炔烃双锡基化的聚合物基单原子催化剂[70]。该单原子催化剂聚合物载体基于单体a23,a23 特殊的结构和电子性质使得催化剂具有很高的活性和选择性。值得注意的是,该作者还指出钯前体对催化剂中金属的分散和催化剂结构均有影响,Pd(PPh3)2Cl2的18 电子结构对钯的单位点分散至关重要。
对于一些水相用催化剂或基于亚磷酸酯配体的聚合物催化剂,其亲疏水性会对催化剂的活性或稳定性产生影响[71-73]。雷以柱等[72]通过改变共聚单体a21 与b17 的比例调控聚合物的亲水性,实现了对聚合物基催化剂在水相中催化Suzuki-Miyaura 偶联反应活性的调控。肖丰收团队[71,73]用亚磷酸酯配体单 体phosphite-tBu (a34) 制 备 了 多 孔 聚 合 物Phosphite-POP,其多孔结构赋予其超强的疏水性(与水的接触角达到152°)。Phosphite-POP 基铑催化剂相较于均相催化体系Rh/phosphite-tBu 具有更高的活性和稳定性。基于a35 和a36 的多孔聚合物催化剂也存在类似的规律,较其相应的均相催化体系活性高。
除了钯和铑,其他金属化合物如AuCl[74]和[Cp*IrCl2]2[75]也被用于与基于3V-PPh3(a21)的聚合物合成多相催化剂。
本部分单体结构、组分、物理性质(比表面积、孔体积和孔径)、负载金属类型以及相应催化剂的应用参见图12、表4。
图12 用于烯烃聚合的单膦配体单体及共聚单体

表4 溶剂热合成的含磷聚合物单体物组成、孔结构参数及其在多相催化中的应用(Ⅰ)
4.2 双膦配体聚合物
基于双膦配体能获得高化学选择性和立体选择性催化体系[76-77],科研人员设计了一系列烯基取代的双膦配体单体,用于合成多孔聚合物载体并运用于多相催化。
2012 年,肖丰收团队[78]改性(R)-BINAP 合成了烯基化的单体a37,经溶剂热聚合、HSiCl3/PPh3还原成功合成了聚合物PCP-BINAP。该聚合物负载[RuCl2(benzene)]2后应用于β-酮酯的不对称加氢,选择性和ee值分别高达99.5%和99.0%。丁云杰团队[79-80]同样通过改性BINAP 合成了烯基化单体(S)-4,4'-divinyl-BINAP(a38)和(S)-5,5'-divinyl-BINAP(a39),指出4(4')位乙烯基取代的BINAP(a38)中磷原子受聚合物骨架的位阻作用较5(5')位取代的a39 更大,因此基于a39 的聚合物钌催化剂在β-酮酯的不对称加氢反应中能达到更高的转化率和ee值[80]。
2015 年,肖丰收团队[81]分别用单体a40、a41和a42 通过溶剂热、后负载Rh(CO)2(acac)的途径制得了聚合物铑催化剂,并将其用于烯烃氢甲酰化反应。相较a41和a42,基于a40的聚合物骨架灵活性好,其31P 核磁谱图信号峰在甲苯中由无甲苯溶剂时的宽信号峰转变成窄而尖的信号峰,表现出“准均相”的性质。该作者认为正是单体a40柔性链赋予聚合物载体“准均相”的性质,使得其催化活性高于基于a41 和a42 的聚合物催化剂。当然,这并不是说基于a41 或a42 的聚合物载体的催化剂活性就低,例如在硫代苯甲酰胺和异腈类化合物合成噻唑类化合物的反应中,基于a41的聚合物钯催化剂就有着很好的活性[82]。随后,丁云杰团队报道了一系列用于氢甲酰化反应的其他聚合物基催化剂,这些聚合物载体基于烯基化的双膦配体单体a39[79]、a43[83-86]和a44[87-88]。其中,基于a43 与a21 共聚物的铑催化剂在C3、C4烯烃的氢甲酰化反应中表现出很高的活性,醛类产物选择性(化学选择性)高达94.2%,且醛类产物中正异比(区域选择性)高达62.2[84-85]。而在长链的正辛烯的氢甲酰化反应中虽有着高的区域选择性,化学选择性较低(醛类产物占58%左右),相当一部分烯烃转化成了相应的异构烯烃和烷烃[86]。而基于a44 与a21 的共聚物铑催化剂在正辛烯的氢甲酰化反应中有着良好的活性和稳定性,在高区域选择性前提下还有着不错的化学选择性(醛类产物占87%)[88]。此外,基于a44 自缩聚的钯纳米催化剂能用作脱羰催化剂,适用于苯环醛基、杂环醛基甚至是长链醛的脱羰基[87]。2019年,贾肖飞团队[89]合成了烯基取代的双亚磷酰胺配体a45,并用于制备烯烃氢甲酰化的聚合物基多相铑催化剂。该催化剂在正己烯的氢甲酰化中表现出超高的活性(TON 达45.3×104),对醛类化合物的选择性为90.3%且正异比高达49.1。当底物换成正辛烯时,活性略有下降(TON仍有1.6×104),但对醛的选择性达91.3%且正异比为41。同年,肖丰收团队[90]合成了超分子离子对膦配体单体a46 和a47,经溶剂热聚合、负载Rh(CO)2(acac)制成催化剂。该类催化剂能在水溶液中催化正辛烯的氢酯基化反应的进行,转化率达94.7%~96.3%,醛的选择性高达98.0%~98.4%且正异比高达39。2020 年,杨勇团队[91]又合成了新聚合单体a45',基于该单体的聚合物基铑催化剂能用于高碳烯烃的氢甲酰化,产物中醛的正异比高达175。值得注意的是,双膦配体单体常与单膦配体a21共聚,进而负载铑金属用于氢甲酰化反应。其中,双膦配体的螯合作用能提供大位阻环境,而单膦配体能起到稳定铑金属物种的作用。
上述基于a42、a43、a44、a45或a45'的聚合物铑催化剂在氢甲酰化中表现出高活性和高选择性,而其膦配体单体对应的双膦配体在均相体系中早已被证明是具有高活性和选择性的配体[92]。这是均相催化体系对基于聚合物基催化剂有指导作用的又一有力证明。因此在均相催化剂被大量研究的领域(如烷氧羰基化反应、氢甲酰化反应、不对称加氢反应等),均相催化体系能为聚合物基催化剂的开发提供很好的指导。
本部分单体结构、组分、物理性质(比表面积、孔体积和孔径)、负载金属类型以及相应催化剂的应用参见图13、表5。
4.3 季盐聚合物
本部分单体结构、组分、物理性质(比表面积、孔体积和孔径)、负载金属类型以及相应催化剂的应用参见图14、表6。

图14 用于烯烃聚合的季盐单体及共聚单体
肖丰收团队在合成多孔聚合物方面做了诸多开创性的工作,如率先报道了溶剂热的方法合成多孔有机聚合物[55],最先报道多孔季盐型聚合物并将其用于多相催化[97]等。丁云杰团队则拓展了含磷多孔有机聚合物基催化剂在多相催化领域的应用,实现了多相催化氢甲酰化的工业应用[20]。在他们的推动下,溶剂热烯烃聚合近十年发展发展迅速,不管
是含磷单体还是不含磷的共聚单体都逐渐变得丰富起来。烯烃聚合能在均相催化体系的指导下设计烯基化的聚合物单体,而且也能引入具有特定功能基团的共聚单体制备多功能的催化剂,在结构调控方面具有巨大的发挥空间。此外,共聚组分(单体及配比)、金属负载量、金属前体以及负载方式(原位加入、后负载等)等都对聚合基催化剂有影响,这一方面使得聚合物基催化剂的可调变性进一步变大,同时也使得聚合物基催化剂的制备更加复杂。作为聚合物基催化剂中起决定性作用的膦配体单体,其合成路径通常比较繁琐且合成条件苛刻,这是溶剂热法聚合物在多相催化领域走向应用的障碍。

表5 溶剂热合成的含磷聚合物单体物组成、孔结构参数及其在多相催化中的应用(Ⅱ)

表6 溶剂热合成的含磷聚合物单体物组成、孔结构参数及其在多相催化中的应用(Ⅲ)
5 其他
其他合成含磷多孔有机聚合物的方法还包括醛胺缩聚、Scholl 缩聚、酚醛聚合、聚吡喃盐的磷代以及多段式聚合等。
5.1 醛胺缩聚
用多元胺和多元醛化合物通过醛胺缩聚的方式是构建COFs 的常用方法[99]。相比其他含磷多孔有机聚合物,长程有序的骨架结构是含磷COFs 独有的特点。这种长程有序包括有序分布的膦配体,特定结构且均匀分布的孔道结构。2019 年,张伟团队[100]使用三(4-甲酰基苯基)膦a56 和对苯二胺b25成功合成了Phos-COF-1(见图15),其孔径主要分布在1.56nm 左右,比表面积为818m2/g。Phos-COF-1 与K2PdCl4、K2PtCl4或HAuCl4在甲醇中混合24h,滴加NaBH4还原后能可控合成平均粒径分别为1.62nm、2.06nm 和1.78nm 的钯、铂和金超细纳米颗粒催化剂。该作者还用K2PdCl4和HAuCl4的混合金属配合物溶液制得基于Phos-COF-1 的铂钯纳米颗粒催化剂,指出该方法的可控性得益于膦配体的配位诱导生长和孔结构的空间限制机制。膦配体与钯、铂或金的前体配位,使得金属离子在Phos-COF-1 均匀分布。金属离子被NaBH4还原成原子后聚集成纳米颗粒,纳米颗粒的生长又受到Phos-COF-1 超细孔结构的限制,最终形成粒径较均匀的超细纳米颗粒。此外,Phos-COF-1 超细孔结构还能在催化剂使用过程中有效限制中金属纳米粒子进一步团聚长大,因此具有较好的重复使用性,重复使用5次金属颗粒只是略有增大。
随后,范以宁团队[101]通过类似的方法获得了P-COF-1和P-COF-2两种含磷COFs。P-COF-1和P-COF-2分别由二元胺单体b25、b26和三(4-甲酰基苯基)膦a56缩聚而来,负载Rh(CO)2(acac)后用于催化苯乙烯的氢甲酰化反应。Rh-P-COF-1和Rh-P-COF-2 首 次 使 用TOF 值 分 别 达2557h-1和2074h-1,重复使用6 次还能维持在2398h-1和2050h-1,具有较好的稳定性。但醛类产物中直链醛与支链醛之比仅为1,区域选择性有待提高。
5.2 Scholl缩聚
Scholl缩聚是芳烃类化合物在酸(AlCl3和质子酸)催化下偶联缩聚合成聚合物的方法。2014年,谭必恩和李涛团队[102]首次将Scholl缩聚应用于含磷微孔聚合物的合成,用三苯基膦a12 和芳烃b13 在AlCl3、质子酸的催化缩聚下合成了肖尔偶联微孔聚 合 物(Scholl-coupling microporous polymers,SMPs),记为SMP-8a(图16)。SMP-8a 负载PdCl2后制成含钯催化剂SMP-8b,在Suzuki-Miyaura 偶联反应中表现出很高的催化活性。Scholl 缩聚合成SMPs简单方便,不过路易斯酸AlCl3的使用增加了后处理难度。SMPs 通过苯环间的共价键直接交联有利于微孔的形成,其固有的大π共轭结构骨一方面赋予聚合物很强的刚性,致使其溶胀性能较差,另一方面也使得其具有发光和半导体性质。
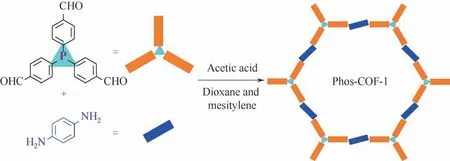
图15 醛胺缩聚合成含磷COFs
5.3 酚醛聚合
2018 年,王忠刚团队[103]通过溶剂热法成功合成了多孔含磷酚醛树脂,将三(4-甲酰基苯基)膦(a56)和2,5-二羟基-1,4-苯醌(b27)溶解在二烷中,90℃加热搅拌1h 制得均匀透明液体,然后将该液体转移至带有聚四氟乙烯内衬的水热釜中,于220℃条件下反应4d制得聚合物体,经洗涤干燥得到含磷酚醛树脂PFN-P(图17)。溶剂热的合成方法使PFN-P 具有很好的多孔性,含有大量的微孔和介孔,比表面积和孔体积分别达775m2/g 和0.72cm3/g。PFN-P 与Pd(PPh3)4的甲苯溶液混合24h得到Pd@PFN-P 催化剂,在Suzuki-Miyaura 偶联反应中表现出很好的活性和稳定性。

图16 Scholl缩聚合成含磷微孔聚合物
上述方法合成的聚合物的单体组成、孔结构参数及其在多相催化中的应用见图18和表7。

图17 溶剂热酚醛聚合合成含磷酚醛树脂
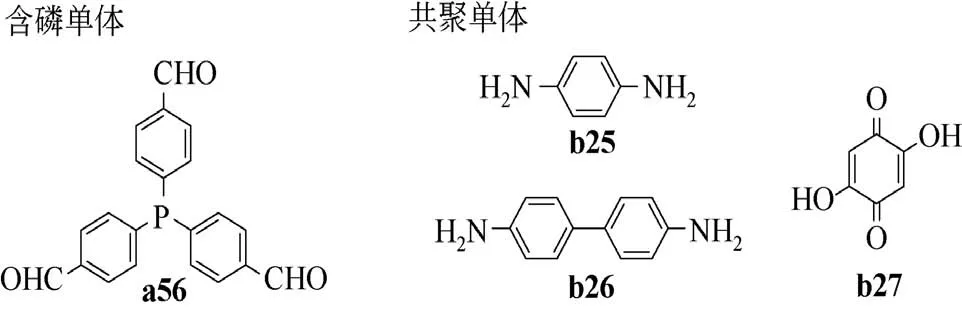
图18 醛胺缩聚及酚醛聚合单体
5.4 聚吡喃盐的磷代
磷杂苯为具有平面构型的芳香性磷化合物,理论计算表明其芳香性为苯环的88%[104]。由于其特殊的空间构型和电子性质,磷杂苯与金属的配位和位阻性质与传统的三价有机膦配体有很大的不同(图19)[105]。因此,磷杂苯聚合物在多相催化中可能具有一些独特的性能。但是磷杂苯很容易被碱[31]或亲核试剂进攻[105]发生化学反应,由磷杂苯单体出发直接合成聚合物的是很困难的。

表7 醛胺缩聚、Scholl缩聚及酚醛聚含磷聚合物单体组成、孔结构参数及其在多相催化中的应用
2019 年,戴胜团队[106]采用“后合成”的策略成功合成了磷杂苯型多孔聚合物(图20),用单体b28、b29 或b30 分别与四(4-乙酰苯基)甲烷缩聚合成聚吡喃盐,再经P(Me3Si)3磷化处理成功制得了磷(Pd@HBPs。 HBPs: honeycomb-like bicontinuous P‐doped porous polymers)[108]、超交联微孔有机纳米棒聚合物基金属纳米颗粒催化剂(M-HMONFs。HMONFs: hypercrosslinked microporous organic nanotube frameworks)[109]、中空多孔有机纳米球基钯催化剂[H-PONs-Pd(PPh3)4。H-PONs: hollow porous organic nanospheres][110]。他们首先通过可逆加成-断裂链转移聚合(reversible addition-fragmentation chain transfer copolymerization,RAFT)合成特定的杂苯型聚合物Phos-POP、F-Phos-POP-1 和FPhos-POP-2。 Phos-POP、 F-Phos-POP-1 和FPhos-POP-2 的比表面积分别为282m2/g、591m2/g和432m2/g,孔径主要分布在0~2nm的范围内。此类聚合物负载RuCl3后用作吗啉的氢甲酰化反应(二氧化碳为羰源)催化剂,结果表明含高浓度氟元素的Ru/F-Phos-POP-2 催化剂表现出相对较高的活性。该作者认为,由于氟对二氧化碳具有亲和性,在催化过程中起到了协同作用从而在三种催化剂中表现出最高的活性。虽然磷杂苯型聚合物丰富了含磷配体的电子性质和配位性质,但磷杂苯结构本身不太稳定,易受亲核试剂、碱等的进攻。这种变化不仅可能发生在磷杂苯聚合物的合成过程中,也可能发生在催化剂的使用过程中。

图19 磷杂苯的位阻性质和配位性质

图20 聚吡喃盐的合成及磷代
5.5 多段式聚合
通过多种聚合手段的组合能设计合成一些具有特殊形貌结构的含磷多孔有机聚合物,可能在多相催化中表现出特殊的性能。黄琨团队通过多种聚合手段,控制合成了一系列基于特殊形貌的含磷多孔有机聚合物催化剂(图21),如微孔有机纳米棒聚合物基钯纳米颗粒催化剂(MONFs-PPh3@Pd。
MONFs: microporous organic nanotube frameworks)[107]、蜂窝状磷掺杂多孔聚合物基钯纳米颗粒催化剂接枝共聚物或嵌段共聚合物;再通过Friedel-Crafts缩聚使含苯环部分交联成形,将聚酯链段刻蚀制成特定形貌的含磷多孔聚合物材料;最后,将金属前体负载于聚合物材料上制得负载型金属催化剂。相较本文中提到的其他方法,多段式聚合法通过对形貌的可控构建增加聚合物催化剂的多孔性,尤其是介孔和大孔。介孔和大孔结构有利于传质,使底物在催化剂体相中能更快的向活性位点扩散。

图21 基于特殊形貌的含磷多孔有机聚合物催化剂
6 结语
不同含磷多孔有机聚合物的制备方法中选用的单体类型是不同的,缩聚反应的难易程度也各不相同,这使得合成的聚合物在骨架结构、溶胀性、规整度、表面性质(比表面积、孔径及孔基分布等)等性质各不相同。偶合缩聚制得的聚合物一般孔径分布较窄,多含微孔或孔径偏小的介孔。锂盐与氯化磷的缩聚反应很容易进行且不可逆,结构无序度高,孔径分布较宽,制得的聚合物多为含微孔、介孔和大孔的多级孔材料。Friedel-Crafts缩聚发生在偶联剂与芳香环之间,苯环上反应位点的不确定性使得聚合物孔结构呈无序分布。溶剂热烯烃聚合为自由基反应,聚合物结构包含大量的柔性烷基链段,通常能表现出很好的溶胀性能。醛胺缩合反应具有一定的可逆性,能合成结构长程有序的COFs。Scholl 缩聚合成的聚合物通过苯环间碳碳键相连,刚性大且主要含微孔结构。磷化聚吡喃盐的方式,通过“后合成”的策略能合成不稳定的磷杂苯类聚合物。多段式的聚合策略,能合成特殊形貌结构的聚合物载体。除了制备方法,缩聚组分(缩聚单体及单体配比)、缩聚催化剂的量、后处理方式等因素对聚合物的物理性质(比表面积、孔结构等)也有影响。此外,这些合成方法中偶合缩聚、Friedel-Crafts 缩聚和Scholl 缩聚等需要金属催化剂参与;溶剂热烯烃聚合、醛胺缩合和酚醛聚合等无需金属催化剂参与;Friedel-Crafts 缩聚和Scholl 缩聚所用单体不需要在膦配体单体引入特定反应位点。每种方法都有其优点和缺点,它们互为补充,丰富了含磷多孔有机聚合物单体类型和结构性质。
稳定性是多相催化剂的一项关键指标,对于聚合物基催化剂来说,影响其稳定性的有多种因素:金属于聚合物中的分布、膦配体浓度、聚合物骨架性质等。金属分布于微孔结构中聚合物骨架对其迁移的阻碍能力比介孔和大孔强,减缓金属间的团聚以及金属的流失。聚合物中磷的浓度越高,其对金属稳定效果越好,对催化剂的重复使用性有一定帮助。在水敏感的聚亚磷酸酯基催化剂中,聚合物骨架强的疏水性对催化剂稳定也能起到关键作用。
金属前体能影响金属在聚合物中的分散形式。总体来讲,以零价金属配合物为前体有利于形成金属纳米粒,而以相对稳定的金属离子的配合物(满足18 电子规则的金属配合物)为前体则有利于金属的原子级分散。对于聚合物基金属纳米粒子催化剂,聚合物中膦配体对纳米粒子在聚合物中的分散度、生长、催化性能均有影响。膦配体能诱导金属前体在聚合物载体中均匀分布,在还原团聚过程中能调控金属颗粒的生长,最后纳米颗粒表面电子性质也受其调控。此外,聚合物孔道也能限制金属纳米颗粒的生长,调控纳米粒子粒径。聚合物基金属单原子或单位点催化剂中,膦配体不仅能调控金属电子性质影响催化剂活性,还能调控其空间位阻控制催化剂的化学选择性和区域选择性,表现出与均相催化体系中膦配体相似的作用。文中有大量的例子说明,在均相催化体系中具有很好活性或选择性的膦配体,其类似结构被引入聚合物载体后能表现出与均相体系相当甚至更好的活性和选择性。均相催化剂体系对聚合物基催化剂体系具有指导意义,比传统无机载体催化剂能更容易得到高活性的催化剂。
前文也提到,大部分合成含磷多孔聚合物的方法都要使用到特定官能基化的膦配体,而官能团化含磷单体的开发和大量合成正是含磷聚合物基催化剂走向应用的一大挑战。还有一点需要指出,本文提到的含磷聚合物基催化剂绝大部分以负载贵金属为主,研究非贵金属催化剂方面的工作很是缺乏。加强对含磷聚合物基非贵金属催化剂的研究与开发,对降低催化剂制备成本、推进工业应用进程具有重要意义。虽然科研人员发现聚合物能对金属纳米颗粒或金属单原子的电子、空间位阻等方面产生影响,但由于缺少研究两者间相互作用的工具,科研人员对聚合物与金属之间作用的理解还很有限。而研究这种关系至少存在两大难点:一是影响含磷聚合物结构的因素多,使得对其可控合成变得十分复杂;二是聚合物催化剂中金属与聚合物配体间的作用是个变化的动态过程。因此还需引入更多动态、原位表征手段或者建立计算模型,以帮助研究人员更好地理解这种作用,进而开发出更高效、稳定的催化剂。
猜你喜欢
作物学报(2022年7期)2022-05-12
储能科学与技术(2022年2期)2022-02-19
气象与环境学报(2021年3期)2021-07-14
药学进展(2021年3期)2021-05-11
药学进展(2021年3期)2021-05-11
井冈山大学学报(自然科学版)(2021年1期)2021-03-05
矿产勘查(2020年3期)2020-12-19
无机化学学报(2020年7期)2020-07-20
物理化学学报(2020年4期)2020-04-24
渤海大学学报(自然科学版)(2020年3期)2020-02-02
