
基于差异共表达分析的肝癌特异性基因的筛选与验证
2021-04-15 03:12岳宇巍王化琨
黑龙江大学自然科学学报 2021年1期
岳宇巍, 王化琨
(黑龙江大学 数学科学学院,哈尔滨150080)
0 引 言
肝癌是我国最常见的恶性肿瘤之一,主要包括两种病理组织学类型:肝细胞癌(Hepatocellular carcinoma,HCC)和肝内胆管细胞癌(Intrahepatic cholangiocarcinoma,ICCA),其中HCC占我国肝癌总数的83.9%~92.3%[1]。据统计,全球每年约有70万例新发肝癌患者,其中大约有35万例肝癌患者在中国[2-3]。
随着DNA测序技术的成熟,基因表达谱数据已广泛应用于癌症研究,如应用基因的差异表达分析(differential expression analysis,DEA)方法和生物信息学工具比较肿瘤和正常组织的基因平均表达水平的差异,挖掘癌症相关的分子标志物[4-5]。DEA是通过识别基因的高表达或低表达筛选潜在的肿瘤标志物,但并没有充分利用微阵列数据,因为它只使用了来自选定基因的信息,而未使用来自整个转录组的信息,且没有考虑它们之间的相互作用[6]。差异共表达分析(Differential coexpression analysis,DCA)可以作为DEA的补充,通过比较共表达网络,认为具有强烈改变的连接性的基因在疾病表型中起重要作用。随着公开转录组学研究的快速积累,结合多个转录组学研究的共表达分析,可以提供更准确和稳健的结果[7]。本文在Marjan等研究5种人类组织的差异共表达和平均表达水平的混杂效应的基础上,结合GEO数据库筛选25个肝组织数据集,根据肝癌发生、发展的3个阶段,构建了新的健康、肝炎和肝硬化的特异性基因对共表达分数集合,并计算了肝癌共表达基因的特异性分数[8]。经验证得到了较好的结果,筛选出肝癌特异性共表达基因,用STRING数据库对这些特异性共表达基因构建蛋白-蛋白相互作用(Protein-protein interaction,PPI)网络,应用Cytoscape软件得到了Hub基因和基因模块,同时,利用GEPIA在线分析工具得到关键基因的差异表达信息及患者生存曲线。利用DAVID在线分析进行GO和KEGG功能富集分析,筛选出与肝癌发生发展相关的关键基因和通路,从基因相互作用角度为肝癌的发病分子机制提供补充和依据。
1 方法
1.1 数据描述
从GEO数据库(http://www.ncbi.nlm.nih.gov/geo/)下载Affymertix人基因组U133 2.0芯片[HG-U133_Plus_2]同一平台号(安捷伦GPL570平台)的25个肝组织基因表达谱数据。首先应用R语言affy包中的函数对数据进行预处理,包括背景校正(Rma)、标准化(Quantiles)、PM校正(Pmonly)和汇总(Medianpolish),然后使用Gemma异常值检测算法去除异常样本[9],再根据样本信息用R语言sva包的ComBat函数移除批次效应,接下来过滤掉平均表达值低的探针,并根据样本信息分成4类(健康、肝炎、肝硬化和肝癌)样本,得到33个基因表达数据矩阵,数据集及样本分类信息见表1。设置每个类别的基因平均表达值在前80%的基因被认为表达。为了进一步过滤肝癌数据集的基因,结合了用R语言处理后的TCGA数据库中肿瘤纯度大于60%的340个肝癌样本,选择平均Counts值在前75%的基因,与GEO得到的肝癌基因取交集得到肝癌基因的研究范围。

表1 GEO数据信息Table 1 GEO data information
1.2 差异共表达分析模型
本文的目的是找到在肝癌组织中的高共表达,而在非肝癌三个组织中低共表达的基因共表达链接。首先在每个数据集中,使用皮尔逊相关系数(Pearson correlation coefficient)为每个数据集建立共表达矩阵。共表达值定义为:Sij={corr(i,j,k) i≠j,1≤k≤33},其中数据集D(k)中基因i和基因j的皮尔逊相关值表示为corr(i,j,k)(这里没有考虑负相关,把负相关定义为0)。对于肝癌的15个数据集中本文选取基因对共表达值在前10%的基因对,并确定在n个数据集中都存在才被认为在肝癌中高共表达并纳入研究范围,用二项分布作为零假设,控制错误发现率(False discovery rate,FDR)为10-4,为肝癌共表达网络选择合理的密度[8]。为肝癌共表达网络的每对链接都计算肝癌特异性分数(Liver cancer specificity score,LCSS),首先定义两个集合(1)和(2),集合(1)定义为在肝癌组织中基因i和基因j的共表达值,集合(2)定义为基因i和基因j分别在健康、肝炎、肝硬化数据集上的平均共表达值,即:

最后特异性分数[8]定义为(3),即:

应用Wilcoxon秩和检验的p值比较了肝癌和其他肝组织中基因对共表达值的秩,Wilcoxon秩和检验可以检验基因对在肿瘤组织和其他三个组织的各个数据集上的共表达值是否有显著差异,结果发现这两种方法结果高度相关。由于p值越小差异越显著,而特异性分数越大差异越显著,于是将p值做负对数变换,发现LCSS与-log10(p-值)相关性为0.88,肝癌特异性分数可以用来表示基因对在肝癌组织上的共表达特异性。应用控制错误发现率的方法为LCSS选择合理的阈值[8],为15个肝癌共表达网络创建了30个随机子集(Random-dataset)作为LCSS的零分布,并且同样计算随机子集中每个链接的LCSS,LCSS-FDR定义为:
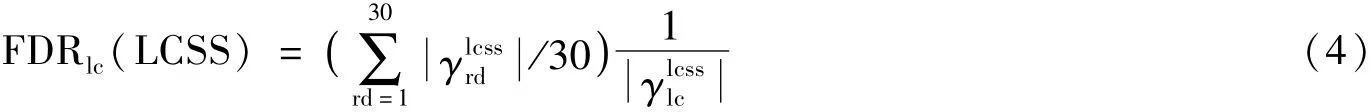
其中γlcssrd是随机子集中大于阈值LCSS的个数,为肝癌组织选择LCSS的阈值控制错误发现率为0.01。
1.3 PPI网络构建和PPI网络挖掘
STRING[10](https://string-db.org/cgi/input.pl/)是已知预测的蛋白质-蛋白质相互作用的数据库,利用STRING数据库构建蛋白质相互作用的PPI网络,再应用Cytoscape软件进行网络的可视化,CytoHubba插件以节点度为筛选条件获得Hub基因,使用MCODE插件获得了重要的基因模块。
1.4 关键基因差异表达验证及生存分析
应用GEPIA[11]数据库对20个Hub基因进行差异表达分析及在线生存分析,差异表达分析验证条件为。生存分析筛选条件为LIHC数据集,置信区间为95%,Hub基因表达量与预后的关系采用Log-rank检验,有统计学意义的差异表示为Log-rank p<0.01或p<0.05。应用The human protein atlas[12](https://www.proteinatlas.org/)得到Hub基因的肝癌预后总结。
1.5 基因模块的富集分析
利用DAVID[13](https://david.ncifcrf.gov/)在线富集分析对模块内的基因进行分子生物学功能(Molecular function,MF)、生物学过程(Biological process,BP)、细胞学组分(Cellular component,CC)的GO功能富集分析,KEGG通路分析,纳入标准为p<0.05。
2 结 果
2.1 利用差异共表达分析挖掘肝癌特异性共表达基因
对预处理后的肝癌数据筛选,最终确定以15个数据集(559个样本和8 759个基因)为肝癌组织研究对象,基因对至少在12个数据集中都存在才被认为是在肝癌组织中高共表达,从而控制肝癌共表达网络密度为0.007,得到了196 589个基因对。LCSS-FDR控制分数的阈值为0.49,阈值过滤后的肝癌特异性网络(Liver cancer specific network,LCSN)包含3 698个基因节点和12 515条边。在LCSN中,选择大于网络平均连通度6的976个肝癌特异性基因作为构建PPI网络的对象。
2.2 肝癌PPI网络的构建和分析
有研究表明,PPI基因对的表达相关性比非PPI基因对的表达相关性更高,在部分PPI基因对中观察到异常高的差异共表达值,并且与高差异表达的基因相比,高差异共表达基因富含更多的肝癌基因[14]。将976个肝癌特异性共表达基因输入STRING数据库构建PPI网络(图1 a),并进一步用Cytoscape软件得到了20个Hub基因和连接紧密的基因模块。
2.3 Hub基因和基因模块的筛选和验证
将上述方法得到的20个Hub基因构建成基因模块(图1b),利用DAVID网站进行KEGG富集分析,结果(表2)确定了9个关键基因富集到重要癌症通路,包括癌症、p53信号、细胞周期、Wnt、PI3k-Akt信号和病毒致癌作用通路。差异表达分析结果显示,有6个基因存在显著的差异表达(图3),生存曲线分析结果(图4)显示,CDK4、RAC1、CHEK1、SPP1、HDAC1和UBE2D1表达的升高和ESR1表达的降低会显著降低肝癌患者的总体生存率(Log-rank P<0.01)。对没有参与这些重要通路的11个Hub基因,通过已发表资料研究最终识别HDAC1、APOB、UBE2D1、ELAVL1、ATG7和MSH2为可能与肝癌发生发展相关的关键基因。

图1 肝癌特异性的976个高连通度基因所构建的PPI网络和枢纽基因模块Fig.1 PPI network constructed by 976 highly connected genes with liver cancer specific and Hub gene module

表2 蛋白-蛋白相互作用网络基因富集分析Table 2 Gene enrichment analysis of PPI network

图2 肝癌特异性对应的蛋白质互作网络中筛选的3个高度互联的模块Fig.2 Three modules with the high interconnection screened from the PPI networks with liver cancer specific
2.4 肝癌模块参与的调控作用
用DAVID富集分析[15]筛选到具有高互联和生物学意义的3个基因模块(图2),其中模块1包含了120个基因,形成了210种相互作用的关系。表2列举了模块富集分析的主要功能,模块1基因富集的主要功能为蛋白质泛素化和泛素蛋白转移酶活性,KEGG通路为泛素介导的蛋白质水解。模块2基因富集的主要功能为脂蛋白代谢过程、脂蛋白颗粒相关功能和多聚腺苷酸核糖核酸,KEGG通路为剪接体和PPAR信号通路。模块3基因富集的主要功能为胞浆,KEGG通路为P13K-Akt和癌症的中心碳代谢通路。

图3 20个Hub基因的差异表达分析结果(红色为肿瘤组,灰色为健康组)Fig.3 Differential expression analysis results of 20 Hub genes(red:tumor group,gray:healthy group)

图4 生存分析结果及患者预后的生存曲线(红色为肿瘤组,蓝色为健康组)Fig.4 Survival analysis results and patient prognosis survival curve(red:tumor group,blue:healthy group)
3 讨 论
随着高通量测序和芯片技术的日益成熟,生成大规模、多组织的基因表达数据已经成为现实。在疾病研究中,以基因表达谱为研究对象、利用生物信息学工具分析的肿瘤研究较多,在众多基因表达数据中挖掘肝癌新型的标志物,为肝癌的诊断与治疗靶点选择及预后判断提供参考具有重要意义。
本研究以差异共表达分析方法通过肝癌发生发展过程的3类数据集(健康、肝炎和肝硬化),准确识别了高差异共表达的肝癌基因对,结合生物信息学工具,对973个肝癌特异性基因进行生物信息学分析,最终得到了3个基因模块和20个Hub基因。在20个Hub基因中,KEGG通路富集分析结果(表2)显示基因CCND1、CDK4、RAC1、CHEK1、RAC2、TP53、ESR1和SPP1主要参与了p53信号、细胞周期、Wnt、PI3k-Akt信号与病毒致癌作用等和肝癌发生发展相关的重要通路,这些参与重要癌症通路的基因已经被广泛研究。除了这些Hub基因,已有研究资料确认,HDAC1[16-19]、APOB[20]、UBE2D1[21]和ELAVL1[22]虽然没有参与这些重要的癌症通路,但是这些基因在肝癌的发生发展过程中起着重要的作用,并且这些基因并没有显著的差异表达(图3),传统的差异表达方法筛选不到这些基因,所以传统的差异表达研究方法并没有全部利用基因表达谱的全部信息,并且生存分析的结果显示CDK4、RAC1、CHEK1、ESR1、SPP1、HDAC1、UBE2D1、ATG7和PRPF8的表达差异会明显降低肝癌患者的总体生存率,这些基因在人类蛋白质图谱的验证中除基因ESR1外,均有对肝癌不利的预后。除了以上基因,MSH2和ATG7基因也可能与肝癌相关,其中MSH2有显著差异表达(图3),并且MSH2和ATG7的高表达也对患者的生存率有显著影响(Log-rank p<0.05),但是这两个基因现在还没有被纳入肝癌研究的靶点。本文提出的差异共表达分析方法有效的识别出了肝癌的关键基因,可以应用在其他类型的多数据集研究,以选择其他复杂疾病的关键基因。本文结果可以作为肝癌差异表达分析研究结论的补充,为肝癌的诊断和治疗靶点选择及预后判断提供参考,多个数据集的联合分析使结论更加具有稳健性。
猜你喜欢
昆明医科大学学报(2020年12期)2021-01-26
天津医科大学学报(2019年3期)2019-08-13
山东医药(2017年20期)2017-07-01
中国医疗保险(2017年5期)2017-05-17
三峡大学学报(自然科学版)(2016年6期)2016-04-16
中国学术期刊文摘(2016年1期)2016-02-13
中国康复理论与实践(2015年10期)2015-12-24
肿瘤预防与治疗(2015年1期)2015-09-26
现代电生理学杂志(2015年1期)2015-07-18
中国当代医药(2015年16期)2015-03-01
