
Eukaryotic microbial distribution pattern and its potential effects on fisheries in the fish reserves of Qiantang River in breeding season*
2021-04-14 05:53HangLAILiZHAOWenYANGReganNICHOLAUSBetinaLUKWAMBEJinyongZHUZhongmingZHENG
Hang LAI , Li ZHAO , Wen YANG , Regan NICHOLAUS ,2, Betina LUKWAMBE ,3,Jinyong ZHU, Zhongming ZHENG,**
1 School of Marine Sciences, Ningbo University, Ningbo 315211, China
2 Department of Natural Sciences, Mbeya University of Science and Technology, Mbeya 53000, Tanzania
3 Department of Food Science and Technology, University of Dar es Salaam, Dar es Salaam 11000, Tanzania
Abstract To examine the eukaryotic biodiversity of aquatic ecosystems in the Qiantang River, China,eukaryotic microbes in the river were investigated using 18S rRNA gene sequencing during the breeding season (July to August 2018). Four distinct distribution patterns (1. Jiande; 2. Tonglu and Fuyang; 3. Jiubao; 4.Yanguan) of the microbial community and their potential eff ects on fishery activities were observed. Results show lower abundances of Dinophyta and Fungi and higher abundances of Cryptophyta and Chlorophyta in Tonglu and Fuyang than those in the other three sections. In addition, the reserves (Tonglu and Fuyang)destabilized the original eukaryotic microbial co-occurrence network. Among all the environmental factors measured, nitrogen (nitrite, nitrate, ammonium), water temperature and total chlorophyll a acted as major driving factors that controlled the eukaryotic microbial distribution. Furthermore, the existence of some algae (e.g., Chrysophyceae, Cryptophytes, and Chlorophyceae) and fungi (e.g., Rhizophydium) in Tonglu and Fuyang was beneficial to juvenile fish growth and water quality, although some detrimental species(e.g., Aphanomyces) needed attention. This study provides further insights into the sustainable protection and utilization of rivers.
Keyword: Qiantang River; fish reserves; 18S rRNA; asymmetric eigenvector maps; molecular ecological network analyses; biological indicators
1 INTRODUCTION
River ecosystems are important parts of terrestrial water that play important roles in the biogeochemical cycling of nutrients (Sun et al., 2014) and fishery production. The increasing human activities in recent years have seriously aff ected river ecosystems,especially those connected to seas (Huang and Huang,2019). The eff ects of human activities could be direct(e.g., sand excavation, dam construction and land reclamation) for urbanization and industrialization(Meyers et al., 2014; Anthony et al., 2015; Andrews et al., 2017) or indirect (e.g., global warming and sea level rising) (Anthony et al., 2015; Chen et al., 2017).Thus, research on the ecology of rivers not only is biologically and geologically important but also has potential applications in river ecological management and environmental protection.
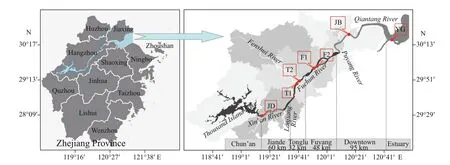
Fig.1 Distribution map of sampling sites
The Qiantang River is the largest river in Zhejiang Province, China, with a drainage area of 44 014.5 km2(Fig.1). It is one of the rarest natural ecosystems and developed as a potential source of freshwater fishery production in Zhejiang (Wang, 1995; Tong, 2001). It has good natural water, a stable water level, abundant bait resources, and sound fish fauna (Tong, 2001).However, overfishing activities have deprived fish resources and destroyed the surrounding environment of the Qiantang River (Su et al., 2014). Therefore,there is an urgent need for quick monitoring and management initiatives to improve the environment of the Qiantang River ecosystem. Some restocking measures have been practiced to conserve the fish resources of the Qiantang River (Zhang et al., 2018b).For example, in 2007 and 2009, Tonglu and Fuyang were selected as provincial-level areas for reservation and restocking, mainly of Chinese carp. The Tonglu and Fuyang sections were selected as conservation areas because they have suitable water quality for fish growth with abundant fishery resources. Furthermore,in 2019 (early March to the end of June), the Qiantang River ushered its first fishing moratorium season and reopened in July, aiming at improving the environment of the Qiantang River ecosystem. The restocking of fish juveniles is one of the most eff ective management activities to improve fish resources in waters, and evaluation of the eff ect of this action on fisheries is essential (Hong et al., 2009). For example, people usually grasp the characteristics of biodiversity by studying the succession of biological resources and environmental factors and the spatiotemporal variation of plankton, benthic organisms and fishes(Zhang et al., 2009). Previous studies on the evaluation of restocking measures in the Qiantang River have mainly focused on the investigation of fish (Zhang et al., 2018b) and benthic organisms (Zhang et al.,2016), while microplankton, including eukaryotic microplankton such as phytoplankton (producers),zooplankton (consumers) and fungi (decomposers),have rarely been investigated.
Phytoplankton, or microalgae, as principal primary producers, constitute more than half of the planet’s primary productivity (Arrigo, 2004). Phytoplankton assembly is exposed to the individual and combined eff ects of various physicochemical and biological factors, such as light, temperature, salinity, pH,nutrients, and grazing. Dramatic variations in phytoplankton communities such as algal blooms may have serious and far-reaching eff ects on the stability of aquatic ecosystems (Huisman et al., 2005).On the other hand, phytoplankton are the main feed for zooplankton and some economic fish, which can aff ect the structure of the zooplankton community and the growth of fish. Phytoplankton are the most important and basic components of the aquatic ecosystem, especially in the food web; thus, studying phytoplankton dynamics is very helpful to further explore the water ecosystem. Zooplankton, including various creatures such as protozoa, crustaceans,mollusks, and their larvae, can be divided into 5 sizes(megalo, >20 mm; macro, 2-20 mm; meso, 200-2 000 μm; micro, 20-200 μm; nano, 2-20 μm). They feed on phytoplankton, organic debris and planktonic bacteria as primary consumers and transfer energy to higher-trophic-level organisms through the food web.Zooplankton are an important feed for many economic filter-feeding fish, aff ecting the material cycle and energy flow in ecosystems and occupying an important position in the trophic level of the waters (Baird et al.,2003). During the main season of juvenile fish growth,zooplankton act as bait organisms and should be widely considered. Like others, the distribution,composition and scale of zooplankton are susceptible to temperature, salinity, transparency, pH and food intake (Tasevska et al., 2012), and some zooplankton can also serve as indicator organisms for assessing the ecological quality of waters (Wen et al., 2011).Therefore, studying the zooplankton community structure is also beneficial for understanding the health status of water bodies and the grazing situation of fish and providing insights into the maintenance of ecological balance in waters. Fungi are often decomposers, parasites, and even consumers and significantly aff ect the growth of fish and the biogeochemical cycling of river ecosystems (Priscu,1995). Previously, microbes were identified using the traditional cultivation method, which has disadvantages of critical cultivation, poor culturability,low resolution, and long duration. Next-generation sequencing (NGS), also known as high-throughput sequencing, which is widely used today, can overcome the preceding shortcomings. Owing to its advantages oflow cost, high throughput, high accuracy, and easy operation, it has been widely affi rmed by researchers in the field oflife sciences. This paper used 18S rRNA gene high-throughput sequencing technology to detect eukaryotic microbes in water.
July to August is the growth conservation period for the juvenile fish in the Qiantang River, and eukaryotic microbes are the main bait for the fish.Therefore, the eukaryotic microbes from July to August were the focus of our study. 18S rRNA gene high-throughput sequencing technology was applied to explore the eukaryotic microbial differences in several major sections of the Qiantang River. This study intends to examine the basic distribution patterns of eukaryotic microbes and water quality parameters of the Qiantang River, explore the main driving factors of the eukaryotic microbial distribution,and investigate the potential relationship between the dynamics of eukaryotic microbial distribution and fisheries.
2 MATERIAL AND METHOD
2.1 Study area and sampling strategy
In July and August 2018, five sampling stations(Jiande, Tonglu, Fuyang, Jiubao, and Yanguan) from upstream to downstream were surveyed in the Qiantang River Basin. As the Tonglu and Fuyang sections are fish restocking zones, they were regarded as the main experimental stations of this research, and each station had two sections; the rest were control stations, with one sampling section each. That is,there were 4 sections of treatment groups, Tonglu 1(T1), Tonglu 2 (T2), Fuyang 1 (F1) and Fuyang 2(F2), and 3 sections of the control groups, Jiande (JD,upstream reference point), Jiubao (JB, downstream reference point 1) and Yanguan (YG, downstream reference point 2) (Fig.1). According to the geographical location, each sampling was performed from upstream to downstream (JD to YG), and each sampling section had three sampling points, which were sampled on both sides and in the center of the river surface (with a depth of 0.5 m). In total, 42 samples were collected (7 sections × 3 sampling points × 2 repetitions). Sample collection, storage,pretreatment, and transportation were carried out in accordance with the methods specified in Surface Water Environmental Quality Standard GB3838-2002.
2.2 Physicochemical parameter analyses
Water temperature, salinity, pH, and dissolved oxygen were recorded in situ using an YSI multifunction water quality detector (YSI 6000, USA) at a depth of 0.5 m, and Secchi depth was estimated by a 0.2-m diameter black-white Secchi disk. The concentration of chlorophyll a was measured in situ using a chlorophyll underwater fluorescence detector(bbe FluoroProbe III, Germany). Ammonium, nitrite,nitrate, orthophosphate, total nitrogen, total phosphorus, and hexavalent chromium were analyzed using an automated spectrophotometer (Smart-Chem 200 Discrete Analyzer, Westco Scientific Instruments,Brookfield, USA) in the laboratory. Chemical oxygen demand was further determined according to the acidic potassium permanganate method. The above analyses were completed within one week after sampling.
2.3 Eukaryotic microbes gathering and DNA analyses
Additional water from the same sites (around 1.5 L) was prefiltered through a 106-μm pore-size sieve and subsequently filtered onto 0.2-μm pore-size polycarbonate membranes (47 mm in diameter,Millipore, Boston, MA, USA) on the sampling day,and duplications were made to preserve. The membranes were immediately frozen at -80 °C until they were needed.
After cutting up the membranes by sterilized scissors, microbial DNA was extracted directly from the membranes using Power Soil®DNA Isolation kits(MO Laboratories, Carlsbad, CA, USA) according to the manufacturer's protocol. Sequentially, the DNA concentration and purity of the extracts were analyzed under the wavelength of 260/280 nm and 260/230 nm using a spectrophotometer (ND-1000, NanoDrop Technologies Inc., Wilmington, DE, USA). The acquired eligible DNA extracts were preserved at-80 °C. PCR was completed under a 30-μL reaction system, and 10 ng of the purified DNA of each sample was used as a PCR template, and the primer pair used 528F (5′-GCGGTAATTCCAGCTCCAA-3′) and 706R (5′-AATCCRAGAATTTCACCTCT-3′) (Peng et al., 2019). The V4 region of the eukaryotic microbial 18S rRNA gene was amplified, and three biological replicates were set for each PCR sample. The PCR conditions were as follows: firstly a predenaturation at 98 °C for 1 min; secondly 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s and extension at 72 °C for 30 s; finally extension at 72 °C for 5 min. The PCR products were then detected by operating electrophoresis on 2% agarose gel, and were extracted again using AxyPrep PCR Cleanup Kit (AXYGEN, Life Science Research). Furthermore,the PCR products were purified by using PCR fragment purification kits (Takara Biotech, Japan).After that, we quantified the purified products using Qubit dsDNA HS Assay kits (Invitrogen, Life technologies) with the Promega QuantiFluor system and picked out those whose concentration was exceeding 2 nmol/L. Finally, moderate equimolar products were degenerated to single streams by sodium hydroxide and were sequenced on the Ion S5TMXL single-end sequencing platform (Thermo Fisher Scientific - CN).
2.4 Processing of original sequencing data
Single-end reads of 303-308 bp (average length of a single sample) were generated and assigned to samples, and the primer sequences were cut off . These matched raw FASTQ data were then processed using the Quantitative Insights Into Microbial Ecology pipeline (QIIME 1.9.0, http://qiime.org/tutorials/tutorial.html). The chimera sequences of the raw reads were detected and truncated by USEARCH 61 to obtain clean reads. Subsequently, phylogenetic types were identified by UCLUST, and sequences were assigned to operational taxonomic units (OTUs)at a 97% similarity level (Edgar, 2010). The most abundant sequences of each phylogenetic type were screened as representative sequences and taxonomically assigned to the SILVA 119 database by PyNAST. We obtained 1 342 019 clean reads and 21 147 to 41 758 reads per sample (31 953±4 473,mean±standard deviation) after quality control,yielding 8 844 original OTUs. To minimize random errors, OTUs that accumulated in all samples at a number below five (perhaps some sequencing errors,approximately 0.001% of the total) (Bokulich et al.,2013) and that were not affi liated with eukaryotes were also removed from the dataset. Additionally, all samples were rarefied to 20 500 sequences to normalize the unequal sequencing depth. Finally,3 407 valid OTUs were selected, and the number in each sample was used to represent species richness.Microbial alpha-diversity indices (Shannon diversity index, observed species richness index and Pielou J evenness index) and the beta-diversity index (Jaccard distance) were computed in the R environment (http://www.r-project.org).
2.5 Statistical analyses
Before ecological analyses, OTU data were Hellinger transformed, and the environmental parameter data were normalized to improve normality and homoscedasticity. Nonmetric multidimensional scaling (NMDS) based on the Jaccard distance of OTUs was applied to visualize overall differences in microbial community structure among sites. The reliability of NMDS was tested by the stress value(Kruskal’s stress formula), which should be lower than 0.2 to minimize misinterpretation (Clarke and Warwick, 2001). The significance and distinctness of differences among groups were assessed by the R-value of ANOSIM (analysis of similarity).Recognition orders were selected by random forest(RF) classification, and 10-fold cross-validation was also implemented. The RF classifier is associated with its Gini that is an index of RF and represents the feature importance of the species. The first axis length by detrended correspondence analysis (DCA) of OTU data was 3.14, which showed a more linear model.Major environmental factors were selected forward and verified with Monte Carlo permutation tests(permutations=999). Then, environmental factors with strong collinearity were also omitted according to the maximum variance inflation factor (VIF).
Asymmetric eigenvector maps (AEMs) (Blanchet et al., 2008), unlike the commonly used principal coordinates of neighbor matrices (PCNM), were applied to directionally quantify the provincial patterns potentially contributing to the community structure. That was, first designing a special tree-like node-edge model (Blanchet et al., 2008) from upstream (JD) to downstream (YG) (namely, those involved from the upper were one, otherwise zero),second transforming their longitude and latitude to Cartesian coordinates (unit=10 000), third weighing their inverse number of squared Euclidean distances,and finally constructing the eigenvector using the function “aem”. PCNM was also applied for comparison. The variation partitioning approach(VPA) was applied to measure the relative importance of spatial and environmental factors on structuring the microbial community. Partial redundancy analysis(pRDA) and partial Mantel tests were applied to clarify the pure correlation between the microbial community and environmental factors. All ecological analyses were performed in the R environment unless otherwise indicated.
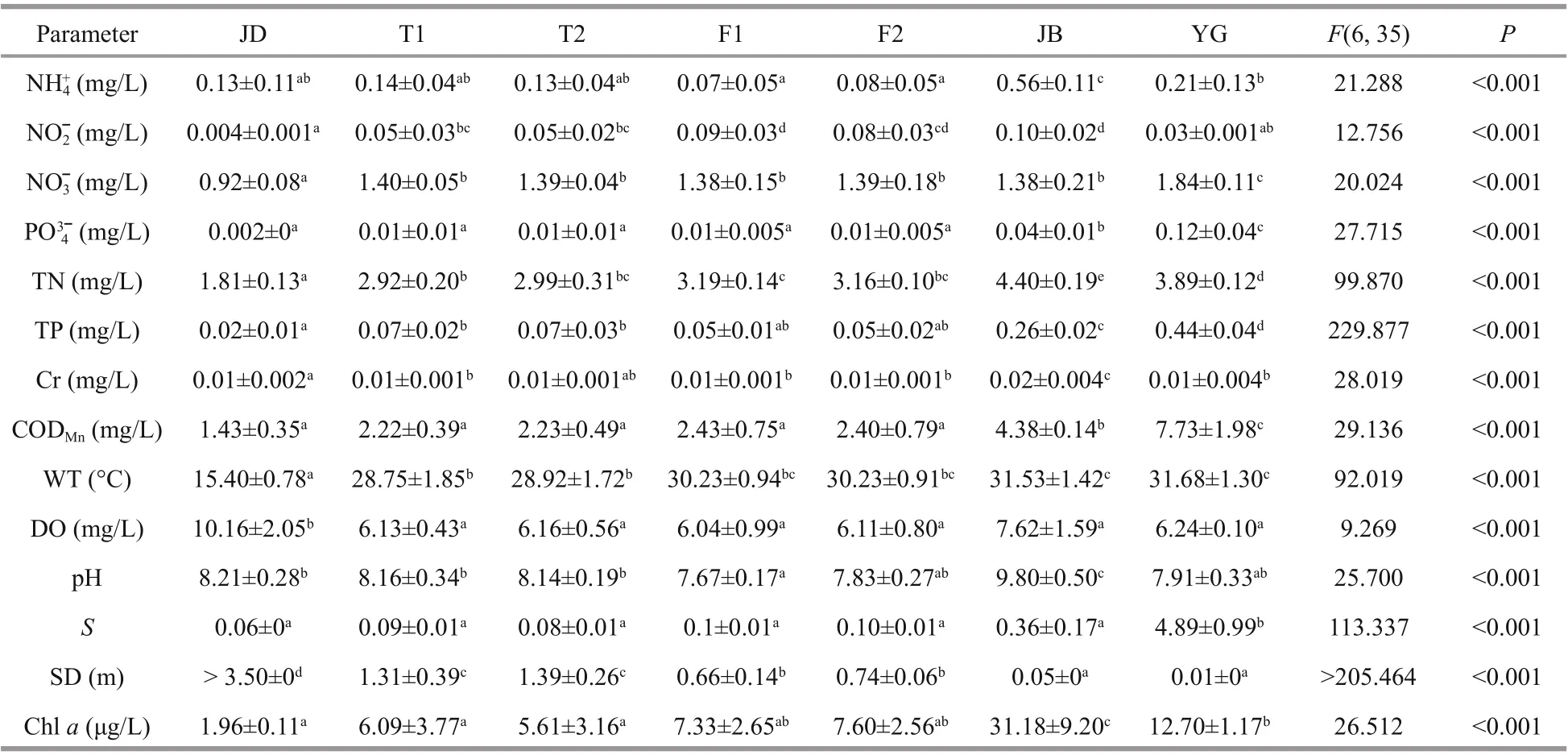
Table 1 differences in basic environmental parameters along the river
Molecular ecological network analyses (MENA)(http://ieg2.ou.edu/MENA) (Deng et al., 2012) based on Pearson correlation matrix and threshold approach of random matrix theory (RMT) were applied to reveal the potential microbial interactions. The topological properties of each network can also be calculated to evaluate their reliability and provide their comparability. Generally, 100 randomly rewired networks will be generated for each empirical network, and network indices are calculated for each randomized network. It is usually ecologically meaningful when the empirical network indices are beyond the randomized network indices.
One-way ANOVA followed by Duncan’s multiple comparisons was performed by SPSS 17 to compare variations in the water quality parameters and alphadiversity indices among sites (the significance level was selected as 0.05).
3 RESULT
3.1 Physicochemical conditions
One-way ANOVA showed that there were significant differences in all environmental variables measured among the sampling sites ( P <0.05, Table 1).In general, nitrite, nitrate, orthophosphate, total nitrogen, total phosphorus, chromium, chemical oxygen demand, water temperature, salinity and total chlorophyll a distributions were minimal at JD and top-down increased gradually via TL (T1 and T2) and FY (F1 and F2), finally peaking at JB or YG, but transparency varied oppositely (Duncan’s test,Table 1). The YG section showed the intricate and irregular variance of the environmental factor itself.
3.2 Distribution patterns of eukaryotic microbial abundance
The NMDS plot clearly revealed the global diff erentiation of eukaryotic microbial community structure (EMCS) among groups (stress <0.1, Fig.2).Its corresponding ANOSIM results ( R=0.918,P=0.001) indicated a high correspondence between TL and FY and their great distinctness against JD and YG and slight distinctness against JB.
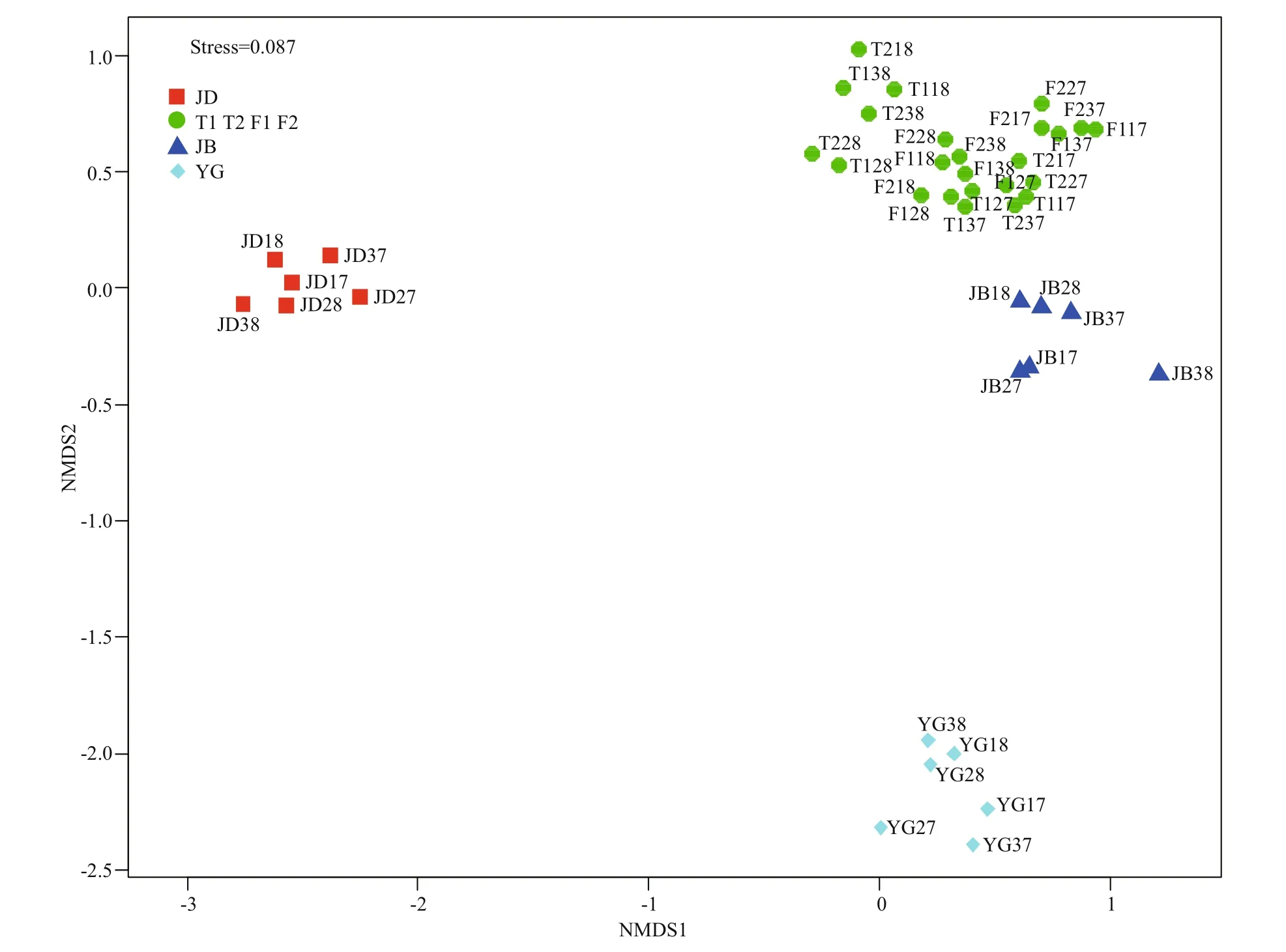
Fig.2 Non-metric multidimensional scaling plot of microbial community based on Jaccard distance in the river
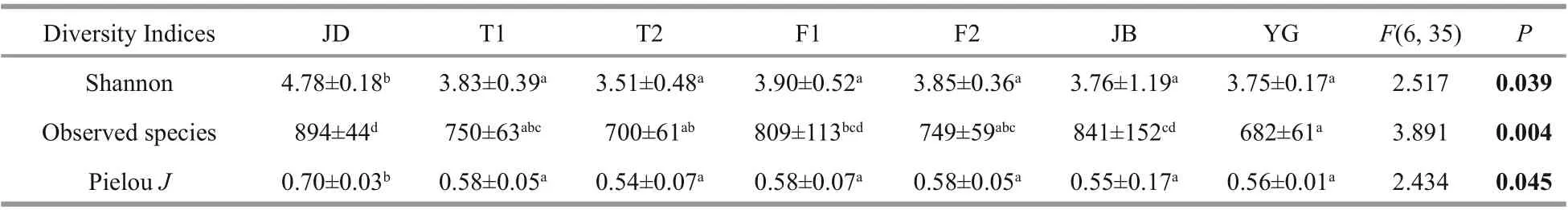
Table 2 differences in microbial alpha-diversity along the river
One-way ANOVA showed there were significant differences in three alpha-diversity indices among diff erent sites with the highest indices in JD ( P <0.05,Table 2).
At the phylum level, Phaeophyta (Ochrophyta),Metazoa, Ciliophora, Cryptophyta, Chlorophyta,Heterokontophyta (Stramenopiles) Other, Dinophyta and Fungi dominated the microbial community across these areas of the Qiantang River (Fig.3), totally accounting for 92.4% (JD), 98.1% (T1), 97.9% (T2),97.8% (F1), 98.2% (F2), 98.1% (JB) and 92.1% (YG)of each section’s OTUs on average. Specifically,Dinophyta hardly existed in TL (0.43%), FY (0.44%)and JB (0.25%) but was relatively more abundant in JD (4.84%, dominated by 68.71% of Peridiniphycidae)and YG (17.17%, dominated by 96.72% of Peridiniphycidae). It was remarkable that up to 31.14%of Gymnodiniphycidae made up the Dinophyta in JD compared to YG (only 1.45% of Gymnodiniphycidae).Similarly, fungi were significantly more abundant in JD (5.77%), JB (2.99%) and YG (4.26%) than in TL(0.62%) and FY (0.89%) (Duncan’s test). In addition,Charophyta had a relatively higher abundance in the JD section (3.36%) than in the other sections (0.04%-0.10%). However, Cryptophyta were observed to be higher in TL (10.63%) and FY (15.89%) than in JB(2.94%) and YG (4.43%) (Duncan’s test); Chlorophyta were detected to be more abundant in TL (9.69%) and FY (13.99%) than in JD (3.86%), JB (5.64%) and YG(4.40%) (Duncan’s test).
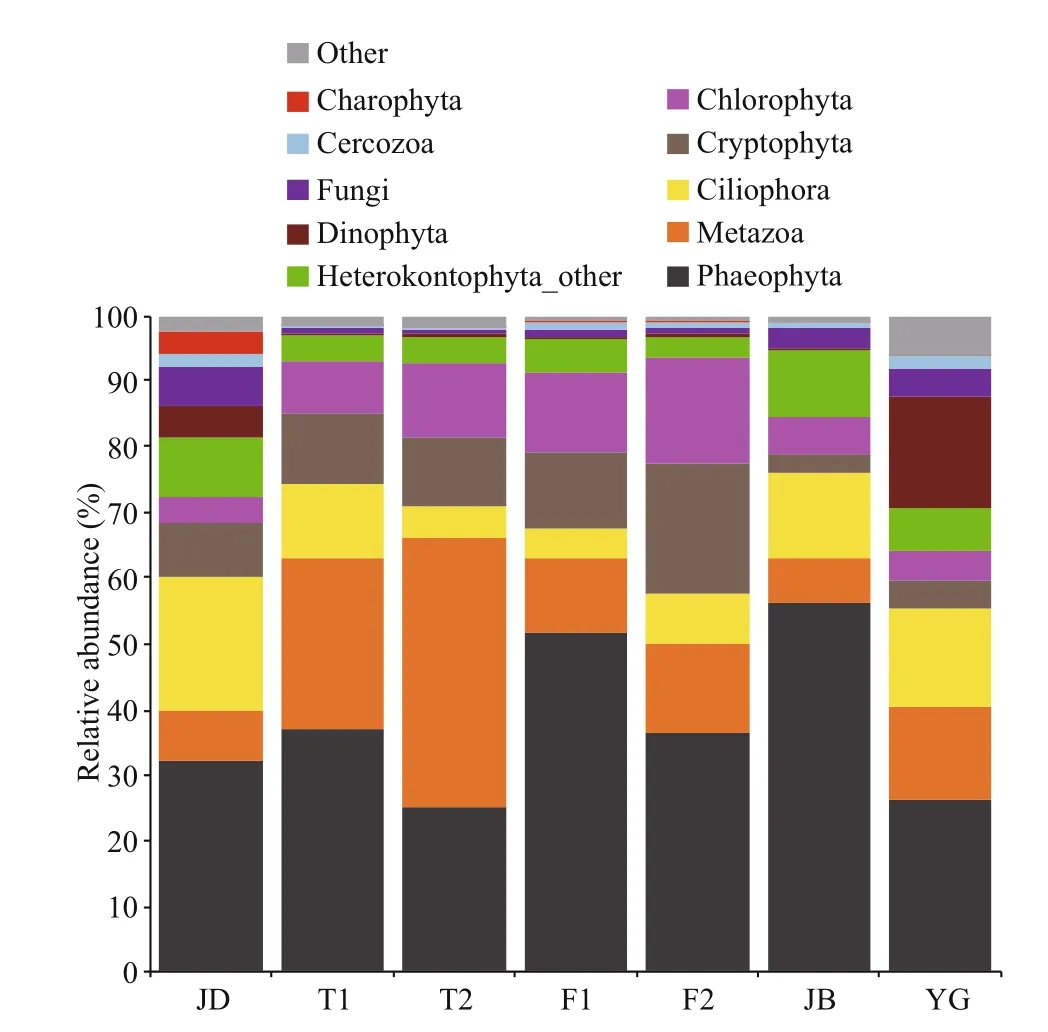
Fig.3 The relative abundance of dominant eukaryotic microbes in the river at the phylum level
The 20 most valid recognition orders of eukaryotic microbes were selected from 379 orders based on the top Gini of RF classification, which further validated the least error of zero by cross-validation. The 20 orders were displayed by heatmap based on their average relative abundance (square root transformed)(Fig.4). It can be seen from the map that the eukaryotes between the left four (TL and FY) and the right 3 (JB,JD and YG) were significantly diff erent (Fig.4). The three top orders ( Coscinodiscophytina,Chytridiomycota, and Peridiniphycidae) may act as biological indicators of JD, JB, and YG, while the four bottom orders ( TKR07M. 92, Teleaula,Hafniomonas and Tetraselmis) may act as biological indicators of TL and FY (Fig.4).
3.3 Environmental determinants of EMCS constrained by geographic factors
Six sets oflinearly independent spatial eigenvalues were generated by AEM. This AEM model could explain the variation in the EMCS by up to 43.7%( F=6.30, P=0.001). Permutation tests by axes showed that only the first four axes were significantly meaningful ( F=14.11, 11.58, 6.24, 4.79, P=0.001,permutations=9 999).
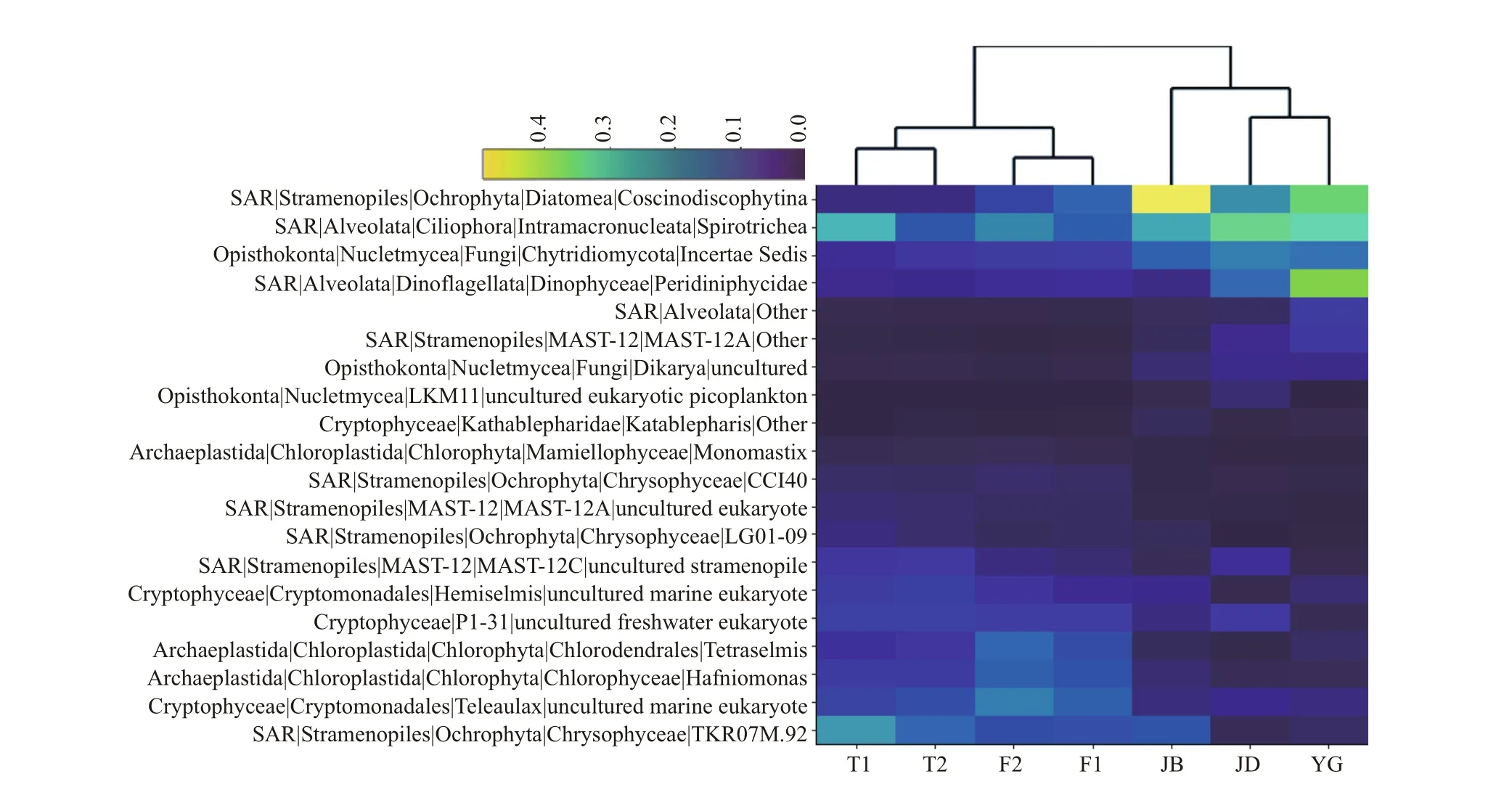
Fig.4 Heatmap of the top 20 microbial orders selected by random forest
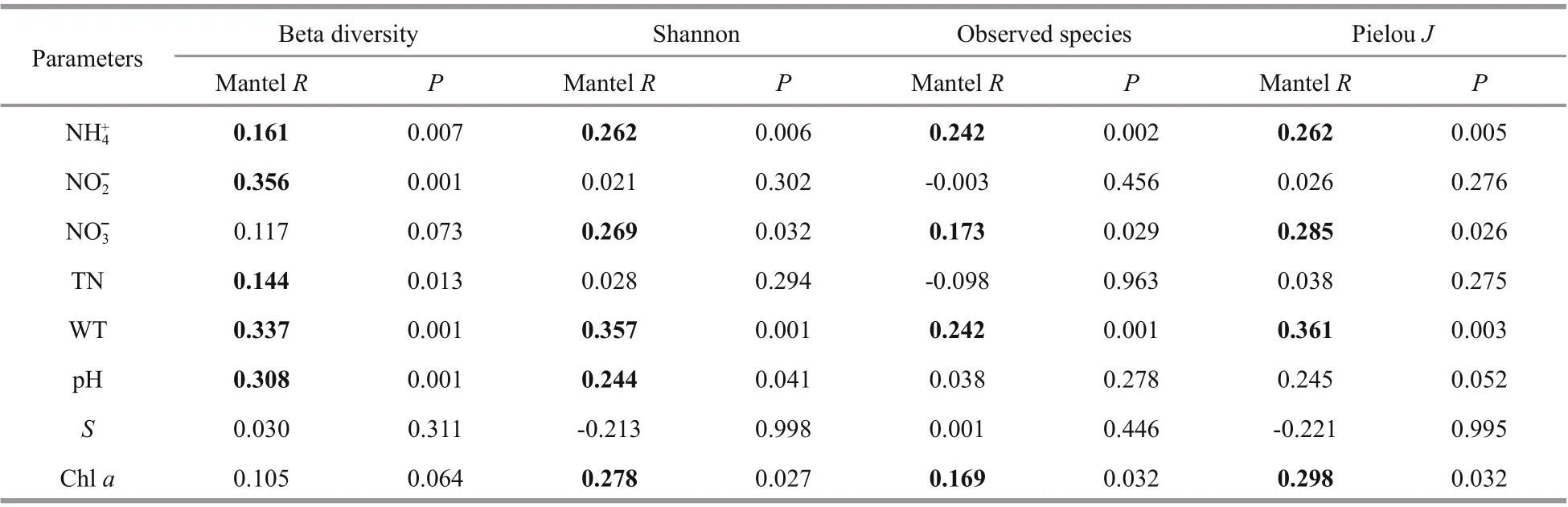
Table 3 Partial correlation between microbial community beta-/alpha- diversity and environmental parameters in the river
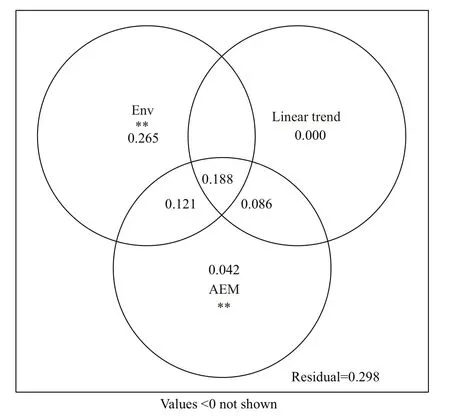
Fig.5 Variance partitioning plot of microbial community structure in Qiantang River
Before the variance partitioning procedure,principal components of environmental factors and the spatial model were extracted as mentioned. Eight environmental factors were retained from 14, namely,ammonium, nitrite, nitrate, total nitrogen, water temperature, pH, salinity and total chlorophyll a, all of which were significantly meaningful by Monte Carlo permutation tests ( P <0.05, permutations=9 999)and explained the EMCS variance by 57.4% (adjusted R2, F=7.91, P=0.001). However, the eigenvalues of the AEM model and linear trend factors were all retained. The final variation partitioning analysis showed that the environmental and spatial variables could explain the variation of the EMCS by 70.2% in total (Fig.5), and only 29.8% was unexplained. Pure environmental eff ects explained the EMCS variance by only 26.5% ( F=4.89, P=0.001) from 57.4%(jointly). The remaining 30.9% explained by environmental factors was spatially constrained. In addition, the pure spatial eff ects could explain the variance by 12.8%, but the linear trend took none ofit without the AEM model.
Partial Mantel test and pRDA (spatial variables as concomitant variables) were opted to reveal the pure relationship of the main environmental factors and EMCS. Permutation tests of partial RDA by axes showed that the first five axes were significantly meaningful ( F=14.96, 10.64, 4.61, 3.26, 3.04,P=0.001, permutations=9 999). The ordination biplot(Fig.6), accounting for 70.1% of the five axes, showed that the variation in the EMCS was intensively correlated with these environmental variables.However, there were no significant correlation in the samples of JB and YG with the environmental variables. In particular, total nitrogen, nitrate, nitrite,ammonium and total chlorophyll a could strongly shape the EMCS of the TL and FY sections in various directions.
The partial Mantel test further validated the pure correlation of the eight main environmental factors and EMCS (Table 3). The beta diversity showed the highest positive correlation with nitrite, water temperature and pH ( P <0.05), while three alphadiversity indices all showed significant positive correlations with water temperature, ammonium,nitrate and total chlorophyll a ( P <0.05).
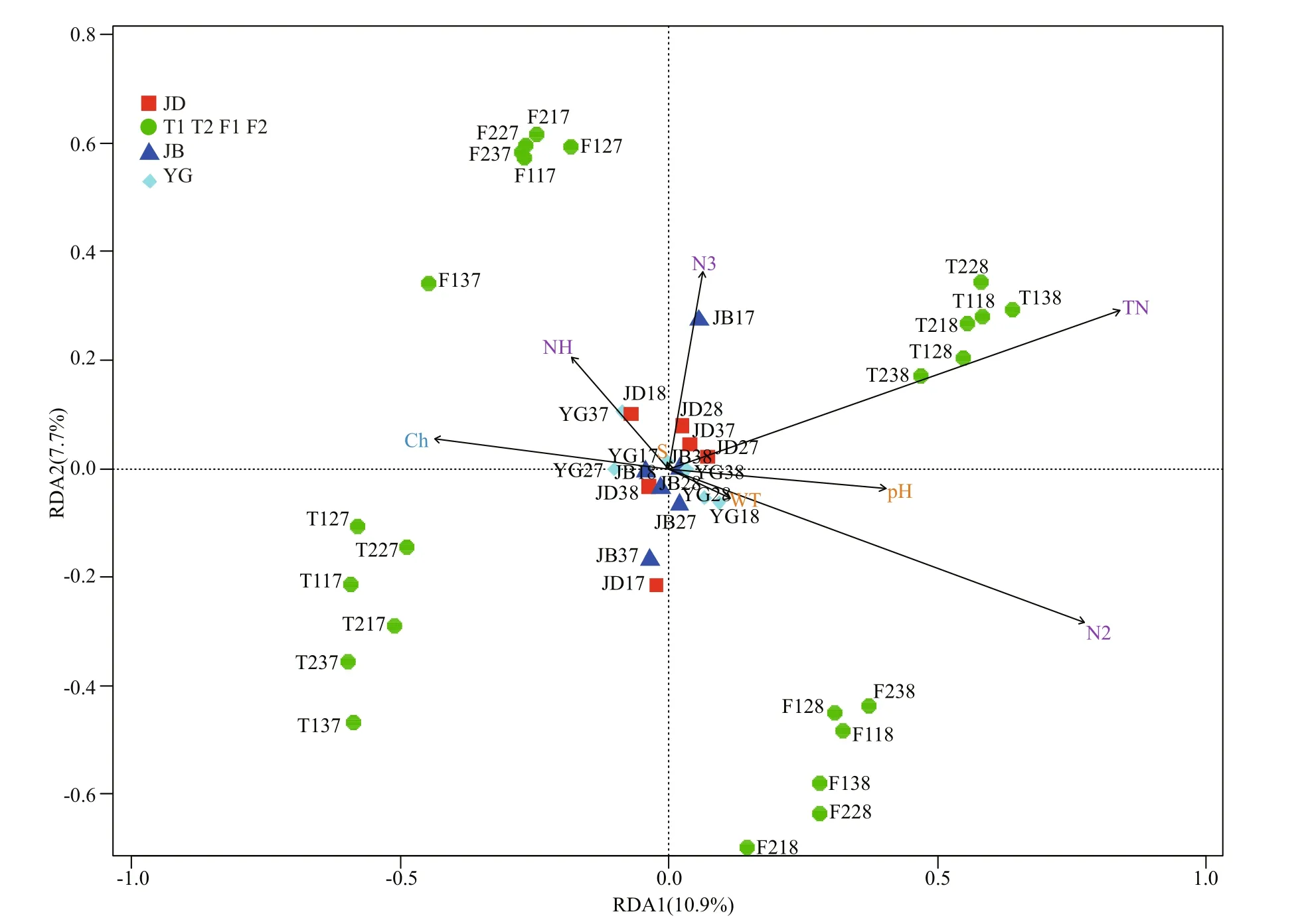
Fig.6 Ordination biplot using partial redundancy analysis showing the pure variations of microbial community constrained by environmental factors in the river
3.4 Topological dynamics of the networks
We chose the class level for simplifying the overcomplex whole networks. As TL and FY were highly similar (Figs.2 & 4), we simply divided all classes into two groups (TL/FY and JD/JB/YG) to highlight the network difference. The network structures and properties of the two networks diff ered clearly (Supplementary Figs.S1 & S2, Table 4). The network of TL/FY was made up of 78 classes, 543 positive and 472 negative links, and 26.0 of average degree; the other was made up of 89 classes, 435 positive and 469 negative links, and 20.3 of average degree (Table 4). Notably, the network of TL/FY possessed fewer nodes but more links than the other,indicating stronger and more complex interspecies interactions. This pattern was also verified by key network topological properties, including more links,a greater degree and higher geodesic effi ciency, and a shorter path distance in the network of TL/FY than the other (Table 4). In addition, nodes with a maximum degree were Chytridiomycota in the network of TL/FY (42) and FV18-2G7 in the network of JD/JB/YG(44), which seemed to be keystones. Interestingly,there were more positive links in the TL/FY network than in the other network (Table 4).
4 DISCUSSION
4.1 Directional spatial modeling
River connectivity and directionality are important for the spread of aquatic organisms (Brown and Swan,2010; Liu et al., 2013a), especially the microbes with smaller size (Chen et al., 2019). Most studies on the metacommunities of the river have used interterrestrial distances among sites to simulate spatial processes without considering connectivity and directionality between river points, such as using the PCNM or distance decay models (Liu et al., 2013b; Zhang et al.,2018a; Isabwe et al., 2019). In this study, AEMs wereused to model the spatial relationship of each river section. The AEM spatial model explained the total variation of the EMCS by 43.7% (adjusted), compared to no more than 35.6% (this value has already exceeded the full model) by the PCNM model ( F=8.57, P=0.001).This result indicated that the AEM model was more suitable for this study and illustrated the importance of considering directionality in the spatial modeling process during water ecology research (Blanchet et al.,2008). In addition, the linear trend variable referred to the coordinates in the Cartesian coordinate system,which cannot explain the community variation alone in this study (Fig.5). This was diff erent from the study on the planktonic eukaryotic microbes in the eastern Zhejiang coast (Zhang et al., 2018a). One possible explanation was that the sampling sections were linearly distributed rather than radially distributed and that the sections were not dense enough in our study.AEM is superior to PCNM or MEM (Moran’s eigenvector maps) in many cases (Vrebos et al., 2017)and has been successfully applied to river ecology(Wan et al., 2015; Isabwe et al., 2018). Thus, increasing reports are considering directionality during spatial modeling in ecology (Zhao et al., 2017; Zorzal-Almeida et al., 2018; Chen et al., 2019).
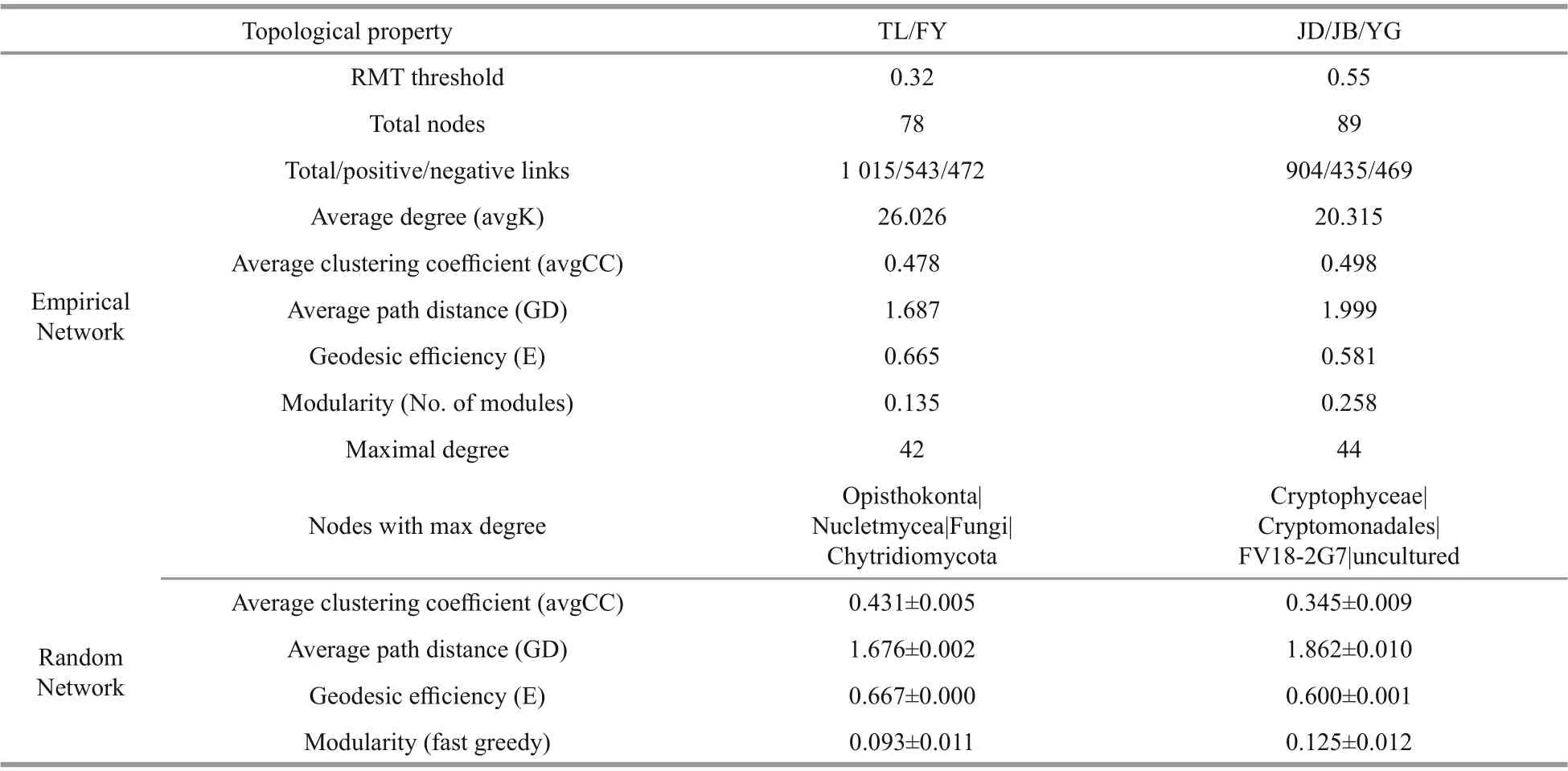
Table 4 Topological properties of the two microbial networks at the class level
4.2 Microbial community structure aff ected by environmental and spatial variables
In this study, the EMCS was significantly correlated with nitrite, nitrate, ammonium, water temperature and total chlorophyll a (Table 3), suggesting that these factors may have the most profound impacts on the EMCS of the Qiantang River. Nitrogen is an essential nutrient for phytoplankton and zooplankton,and a strong correlation between the concentration of nitrogen and the biomass of phytoplankton and zooplankton has been well documented (Zhang et al.,2018a; Shetye et al., 2019; Zhang et al., 2019).However, nitrogen is the most common but limited macronutrient in water, which could limit the primary productivity of water by regulating phytoplankton growth (Abell et al., 2010). Meanwhile, nitrogen can indirectly promote zooplankton growth, as a high level of nitrogen helps phytoplankton grow, which could provide more resources for filter-feeding zooplankton to thrive and reproduce (Zhang et al.,2019). In this study, partial RDA results also provided affi rmative evidence on this (Fig.6, e.g., ammonium,nitrite, nitrate and total nitrogen).
Water temperature is generally thought to be a vital basic environmental factor aff ecting EMCS (Liu et al., 2013b; Duarte et al., 2019) and prokaryotic community structure (Hu et al., 2018) in direct and/or indirect ways. Water temperature usually results in large gradient changes in physicochemical factors of aquatic ecosystems (Lv et al., 2013; Liu et al., 2015),which could easily cause a series of chain reactions of the EMCS. High temperature coupled with elevated nutrient concentrations, for example, can increase the occurrence of phytoplankton blooms (e.g., the JB section of this study) and influence the density of potential pathogens and the activation of virulence factors (Kinnula et al., 2017). However, as the sections we studied were too typical to form huge differences in water temperature, only a weak constraint by water temperature could be found according to the partial RDA plot (Fig.6) compared with the nitrogen above.
Chlorophyll a concentration could be a measurement of phytoplankton biomass (Zhang et al.,2019), but it could not fit well in this study. This is likely because 18S rRNA gene sequencing technology has some methodological limitations; for example,cyanobacteria (also a kind of phytoplankton) were classified as prokaryotes in high-throughput sequencing. Furthermore, our bbe FluoroProbeIII detector may perform poorly or insensitively when coping with epinephelos water, as the chlorophyll a data seemed slightly exaggerated in JB and YG(Table 1). However, both correlation and partial RDA with the EMCS showed a considerable constraint by chlorophyll a. Some studies showed that a higher chlorophyll a concentration caused a concurrent increase in microbial density (DeBoer et al., 2016),but this was inconsistent with our study probably due to the complexity of the eukaryotic microbial community or the eff ects of the predators (e.g. juvenile fish) which feed on zooplankton. Moreover, a higher concentration of blue-green algae (i.e., cyanobacteria blooms) may enhance the bacterial ecological process,leading to increasing the abundance of bacterivores(e.g., Ciliophora and Cercozoa) and the whole eukaryotic microbial community (Xue et al., 2018).However, cyanobacteria blooms could inhibit the growth of zooplankton at times due to the toxins released (Liu et al., 2019). In addition, other detected microbes, such as fungi and aquatic larvae, may also reflect the variance of chlorophyll a concentration,thus aff ecting the EMCS.
Significant eff ects of pH on the beta diversity and Shannon index (Table 3) may be attributed to increased calcification of the plankton’s cytoderm at higher pH (De Nooijer et al., 2009). In addition to these factors, other environmental variables involved only showed weaker or no correlations with the EMCS (e.g., salinity, Table 3). Besides, there were no significant correlation in the samples of JB and YG with the environmental variables (Fig.6), which might be due to the eff ects of the tide, salinity, turbidity or other hydrological factors. Actually, the eukaryotic microbial community is mostly aff ected by stochastic processes rather than deterministic processes (e.g.,environmental factors) in turbulent waters, which needs a further study (Chen et al., 2019).Although the importance of environmental and spatial variables in controlling microbial communities has been recorded (Chust et al., 2013; Zhang et al.,2018a), it is still diffi cult to decide which process is dominant. Our present study also showed severely spatially structured profiles of environmental variables. The spatially structured environmental variations explained most of the EMCS variation(Fig.5), indicating that the local environmental conditions were spatially shaped and conditioned the EMCS. However, there are many unknown factors that we did not measure, which might cause an overestimation of the current variations owing to the limited approach (Isabwe et al., 2019).
4.3 Relationship between eukaryotic microbes and fisheries
The TL and FY sections were both fish restocking and reservation areas of the Qiantang River, and many eukaryotic microbes were important feeds for fish.The microbes in this study had an extensive range of cell sizes. Larger cells, including unicellular organisms (e.g., ciliates, diatoms and dinoflagellates)and multicellular organisms (e.g., rotifers and mollusks), generally possess much higher 18S rRNA copies per unit than those of other microalgae and protists (Gong et al., 2013), which could induce an overestimation of their relative abundance. However,this study focused more on whether specific microbes could be detected, and their relative abundance was a secondary consideration. Rotifers are important members of zooplankton, which are the initial bait of most fish larvae and the main feeds of filter-feeding Chinese carp, so they are essential for fishery production. In this study, rotifers were distributed more in the TL section. The rich and nutritious animal feeds and rotifers can provide adequate material insurance for fish larvae, which is beneficial for fish growth.
Rhizophydium of Chytridiomycetes fungi can eff ectively control the occurrence of cyanobacterial blooms (Frenken et al., 2017; Ortiz-Cañavate et al.,2019). A considerable number of Rhizophydium were distributed in TL and FY in this study, which played a certain role in the regulation of water quality.Healthy water rears good fish. However, some fungi(e.g., Chytridiales) can be saprophytic or parasitic on hosts (e.g., fish and diatoms) (James et al., 2006).Some fungi can even cause epidemics. For example,Aphanomyces of Oomycetes, an opportunistic pathogen, can cause epizootic ulcerative syndrome(EUS), whose mortality rate ofinfection is close to 100% (Saylor et al., 2010). In this study, a small amount of Aphanomyces was detected in TL and FY(but the taxonomic status belonged to the Peronosporomycetes). Therefore, that the fact that this fishery disease may be controlled by biological control methods such as Bacillus sp. strain BC01 ifit occurs in TL or FY requires further study (Foysal and Lisa, 2018).
In phytoplankton, Chrysophyceae have bare cell walls, are rich in eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), and have characteristics of good palatability and rich nutrients. Therefore,they are natural high-quality baits for aquatic animals.In this study, Chrysophyceae are widely distributed in TL and FY (Fig.4). Cryptophytes are indicators of a healthy water quality; the difference in their distribution indicated that the water in the upper reaches of FY was better than that in the lower reaches of JB (Figs.3 & 4). In addition, they are also excellent baits preferred by many filter-feeding fish, especially Chinese carp. In this study, Cryptomonas and Teleaulax of Cryptophytes were widely detected in TL and FY (Fig.4). Chlorophyceae are also important baits for aquatic animal larvae and filter-feeding fish,some of which are beneficial to water quality. In this study, quite a few Tetraselmis, Chlamydomonas and Hafniomonas of Chlorophyceae were detected(Fig.4), which were distributed mostly in TL and FY.In summary, most microbes of TL and FY were beneficial plankton; their presence could improve water quality and provide natural bait, but there were also a small number of harmful microbes that needed attention.
In this study, the environmental factors and the EMCS of TL and FY were highly consistent and significantly diff erent from others (Figs.2 & 4), so we can simplify all the sequences into two categories(TL/FY representing the fish reservations, JD/JB/YG representing the nonprotected areas) to highlight the overall differences in the molecular ecological network formed by diff erent ways of management,although some secondary information might be ignored. The two networks generated also had clear general network properties (Deng et al., 2012), such as being free-scale and of small world (Table 4).Positive links (correlations) in the network may suggest similar ecological functions between the nodes, while negative links may indicate competition or predation relationships (Steele et al., 2011).Although cooperating networks are effi cient, they can also be unstable; while competition may drive ineffi ciencies, it will dampen the unstable cooperation eff ects that can lead to a loss of community members(Coyte et al., 2015); in turn, it shows more stability.This study showed that the proportion of cooperative interactions (positive associations) in the TL/FY network far exceeded the proportion of JD/JB/YG(Table 4), indicating relatively lower stability in the fish reservations (TL and FY).
Another method can evaluate community stability.Interspecies interactions respond directly and instantly within a community when encountering perturbations,so the strength ofinteractions can also determine the stability of an ecological community (Shade et al.,2012). The network complexity (assessed by the network topological properties, Table 4) could be used to infer the strength ofinterspecies interactions(Faust and Raes, 2012). Generally, a more complex community means a more stable ecosystem (Neutel et al., 2007), but the exact relationship between complexity and stability remains controversial (Faust and Raes, 2012). It has been proposed that a successionally complicated network would destabilize a naturally assembling ecosystem (May, 2001),resulting in weaker stability. In this study, a more complex community brought an ecosystem of weaker stability (TL and FY). One possible explanation for this was that anthropogenic restocking behavior might upset the original ecological balance by applying survival pressure on plankton, thus resulting in a decrease in the plankton (nodes) but more intensive associations in the TL/FY network, which means unstable turnover. However, we considered the turnover to be temporary, occurring just during the transition period to a higher stability level. A series of measures would have been implemented, such as fertilization and fish harvesting, to relieve the existence pressure of the plankton. Most likely, soon the fish reservations of the Qiantang River would eventually reach a higher level of stability. These conclusions again confirmed our finding that the reservations destabilized the microbial community.
Topologically, species play diverse roles in an association (Guimerà et al., 2007). Although keystones are usually the minority of an association(based on network topology and modularity), they often play principal roles in response to external perturbations (Deng et al., 2016). For example, in this study, Chytridiomycota (only 0.65% of all sequences)were the keystones of the TL/FY network (Table 4). It has been reported that members of Chytridiomycota can regulate the number of diatoms (Ibelings et al.,2004; Lefèvre et al., 2008; Xue et al., 2018), which was consistent with this study (Supplementary Fig.S1). Pathogens can suppress the growth of phytoplankton that have been infected by specific pathogens; thus, fungal parasitism can be a vital factor controlling natural succession (Ibelings et al., 2004).
5 CONCLUSION
This study demonstrated that the magnitude of differences in the microbial community assembly and alpha diversity diff ered along the selected sections of the Qiantang River, and the distinctness was significantly constrained by spatially structured environmental factors and biological interactions.The distinctions can be significantly categorized into four patterns, namely, (i) JD, (ii) TL and FY, (iii) JB,(iv) YG, perhaps owing to the relative manager’s restocking measures in TL and FY. Among all the environmental factors measured, nitrogen (nitrite,nitrate and ammonium), water temperature and total chlorophyll a acted as major driving factors that controlled the eukaryotic microbial distribution. The existence of some algae (e.g., Chrysophyceae,Cryptophytes and Chlorophyceae) and some fungi(e.g., Rhizophydium) in TL and FY could be beneficial to juvenile fish growth and water quality, although some detrimental species (e.g., Aphanomyces) need attention. In addition, we also identified some potential keystone microbes, indicating their importance in regulating the microbial community along the river.These findings provide valuable information for supplementing the available knowledge on eukaryotic microbial ecology in flowing rivers with contrasting human activities using 18S rRNA gene sequencing technology.
6 DATA AVAILABILITY STATEMENT
The data that support the findings of this study are only available from the corresponding author on reasonable request, due to some limitation.
7 ACKNOWLEDGMENT
We would like to thank the Beijing Novogene Biotechnology Co., Ltd. for 18S rRNA gene sequences. The authors wish to thank the anonymous reviewers for their valuable comments and suggestions, which were helpful in improving our manuscript.
 Journal of Oceanology and Limnology2021年2期
Journal of Oceanology and Limnology2021年2期
- Journal of Oceanology and Limnology的其它文章
- Predicting sediment flux from continental shelfislands,southeastern China*
- Laboratory simulation of dissolved oxygen reduction and ammonia nitrogen generation in the decay stage of harmful algae bloom*
- Development of high-resolution chloroplast markers for intraspecific phylogeographic studies of Phaeocystis globosa*
- Effects ofiron and humic acid on competition between Microcystis aeruginosa and Scenedesmus obliquus revealed by HPLC analysis of pigments*
- Effect of river plume on phytoplankton community structure in Zhujiang River estuary*
- Exploring the sublethal genotoxic effects of class II organophosphorus insecticide quinalphos on freshwater fish Cyprinus carpio