
赤铁矿(100)表面的甲醛吸附特性的反应分子动力学模拟
2021-04-13 03:22姚冠宇李中洲黄正兴唐祯安
仪表技术与传感器 2021年3期
姚冠宇,余 隽,吴 昊,李中洲,黄正兴,唐祯安
(大连理工大学电信学部生物医学工程学院,辽宁省集成电路与生物医学电子系统重点实验室,辽宁大连 116024)
0 引言
氧化铁以纳米粒子和超薄薄膜的形式广泛存在于自然界中,其中赤铁矿是室温下最稳定的铁氧化物之一[1]。由于其表面活性、热稳定性以及自然丰度,赤铁矿正越来越多地用于环境和能源领域中[2-8]。在气体传感器领域,氧化铁用于检测甲醛及其他气体已经开展了许多研究[9-10],但氧化铁对于气体的传感机理尚不清楚。近年来,针对氧化铁的不同表面如(001)[11-13]、(012)[14-16]、(1-12)[17]等进行了广泛的实验和计算研究。在氧化铁的多表面中,(100)表面是常见的伴生面,并且是唯一的非极性面,而表面极性对气体吸附起着重要的作用。基于上述原因,选择赤铁矿(100)表面进行研究。本文通过反应分子动力学方法研究了甲醛在α-Fe2O3(100)表面上的吸附,分别模拟了只有甲醛以及甲醛与空气中气体(氧气、水、氢气)混合的情况,得到了不同温度下的吸附结构和能量,同时计算了不同模型的径向分布函数。
1 模型构建
α-Fe2O3的空间群为R-3C,晶系为六方晶系,晶胞参数如表1所示。使用Materials Studio建立仿真模型,首先建立3×5×5个晶胞构成的超胞模型,截取α-Fe2O3的(100)表面,并在表面增加5 nm的真空层。在氧化铁(100)表面5 Å范围内放置相应的气体分子(1 Å=10-10m),得到最终的计算模型。模型总尺寸为2.517 5 nm×6.86 nm×6.308 13 nm,建立的α-Fe2O3气体吸附模型如图1所示。

表1 α-Fe2O3的晶胞参数

图1 α-Fe2O3和气体混合模型
2 分子模拟方法
2.1 ReaxFF力场
ReaxFF(reactive force field)力场最早是由M. Aryanpour等[18]提出的适用于化学反应系统的大规模分子动力学模拟仿真力场。它利用键长/键序和键序/键能之间的关系来实现键和非键系统之间的平稳过渡,因此,使用ReaxFF力场可以模拟传统势场不能模拟的化学成键、断键现象,更适用于描述气体吸附这类发生化学反应的体系。
2.2 仿真设置
使用LAMMPS软件包进行分子动力学模拟,并使用ReaxFF力场描述Fe、C、O、H原子之间的相互作用。α-Fe2O3(100)表面和气体在吸附之前的初始结构如图2(a)所示。气体混合物的初始压力约为0.76 MPa,时间步长为1 fs,体系自由弛豫。

(a) (b) (c) (d) (e)(f)
(a)在0 ps时的初始状态;(b)~(f)在50 ps时的不同温度下的状态。
将系统的3个方向均设置为周期性边界条件,体系计算采用正则系综(NVT),仿真时间为60 ps。体系分别在300、400、500、600、700 K的吸附结果。同时分别计算体系总的能量以及单独气体和单独氧化铁的能量,可得到体系在该条件下的吸附能。
3 结果分析
将单独水存在时在氧化铁(100)表面的吸附结果与使用量子力学[19]计算得到的结果进行对比,如图3所示。可以看出使用两种方法得到了相同的吸附构型,因此使用ReaxFF可以验证氧化铁(100)表面对气体进行吸附的模拟结果是可靠的。
图3(a)为本文模拟得到的结果,OFe为氧化铁中氧;OW为水中氧;图3(b)为量子力学得到的结果,图中T、D分别为氧化铁中的铁和氧;S、H分别为水中的氧和氢。
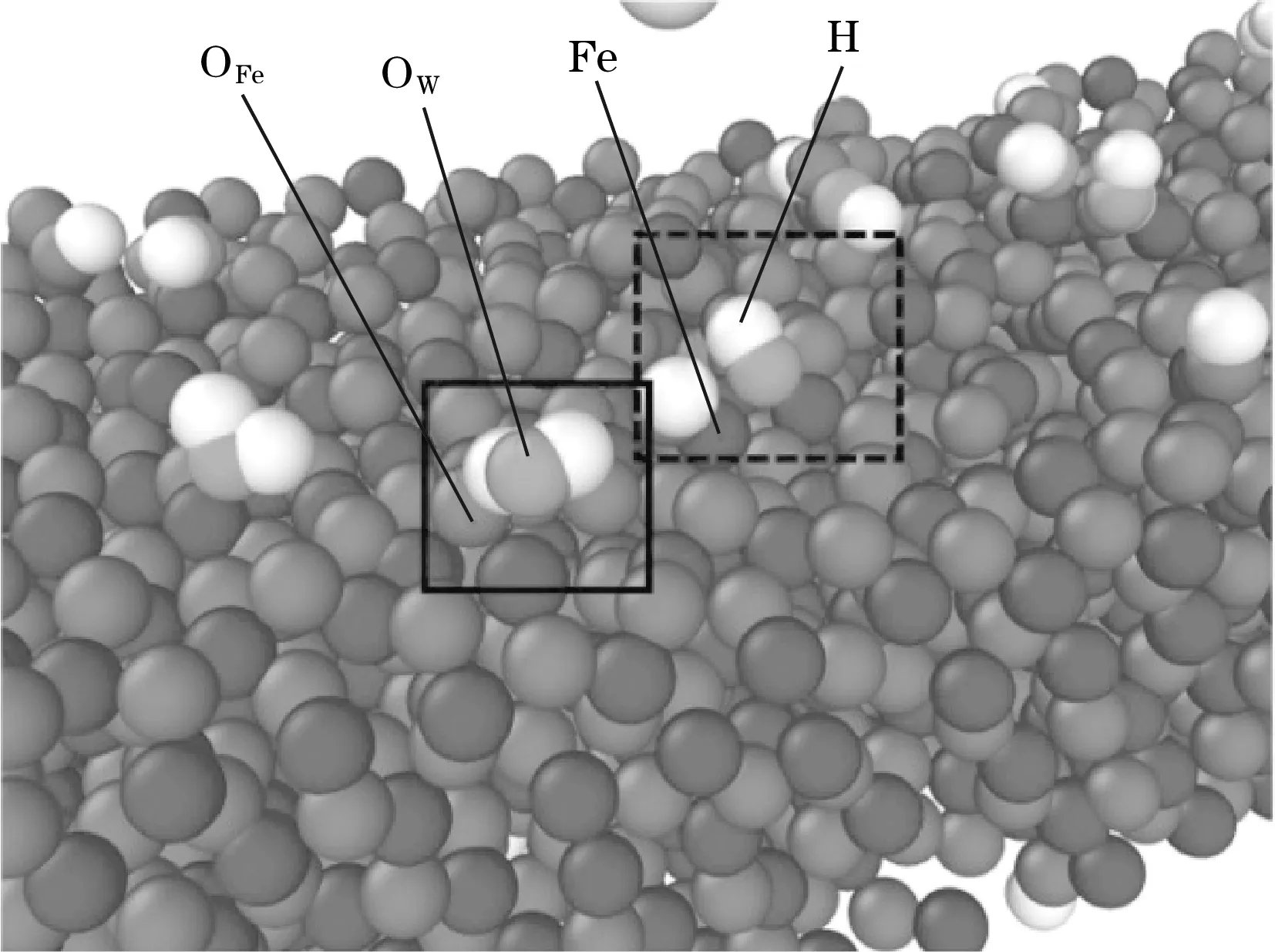
(a)本文模拟得到的结果
气体分子在α-Fe2O3表面上的动态吸附过程通过分子动力学模拟可以清楚地观察到,图2(b)~(f)展示了不同温度下在50 ps时系统的吸附结果。 图4所示的吸附能由式(1)计算得到:

图4 不同温度的吸附能
Ead=Egas/Fe2O3-(Egas+EFe2O3)
(1)
式中:Egas/Fe2O3为吸附后系统的能量,kcal/mol;Egas和EFe2O3分别为吸附前气体和氧化铁的能量,kcal/mol。
在300~700 K范围内,单独甲醛分子的吸附能均为负值,这表明甲醛在任何温度下均可自发吸附在氧化铁(100)表面上。在气体混合物的吸附体系中,分别统计距氧化铁表面0.2 nm范围内的各气体分子的原子数,统计结果如表2所示。在4种气体中,甲醛最容易吸附在氧化铁(100)表面上,其次是水,然后是氢气,最后是氧气。

表2 α-Fe2O3的晶胞参数
氧气对于还原气体在n型SnO2、ZnO上的吸附起重要作用,R. Barik[9]以相同的方式解释了甲醛在氧化铁表面的反应。但是本文的模拟结果表明,氧气对甲醛的吸附没有明显影响,甲醛分子直接吸附在氧化铁(100)表面上,其典型的吸附结构如图5(a)所示。
对比只有甲醛和甲醛与水混合气体的吸附结果(图5),水的存在使甲醛在氧化铁(100)表面上的吸附更强。2种情况下甲醛中氧原子与氧化铁中的铁原子之间的径向分布函数结果如图6所示,可看出当有水存在时甲醛中的氧原子与铁原子的结合更紧密。
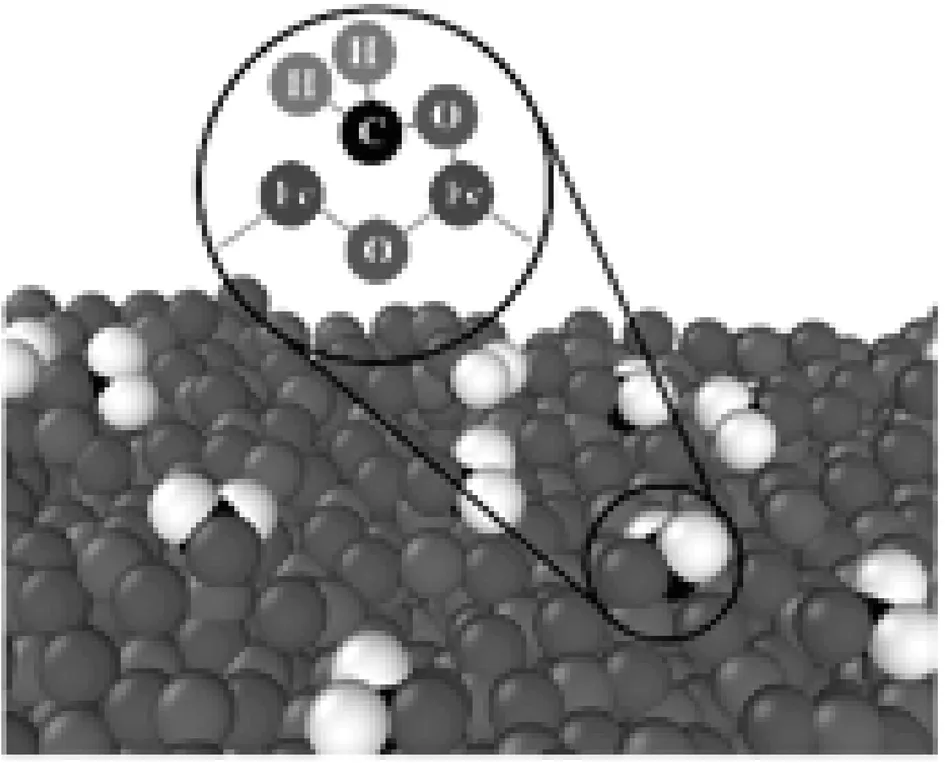
(a)只有甲醛分子

(a)只有甲醛分子
对于含水的体系,其吸附能在600 K时最低,在此温度下发现了新的甲醛吸附结构:当甲醛靠近水分子时,甲醛将分解为一个CH2基团和一个O原子,并且O原子从水中带走一个氢原子与水分子形成羟基基团,吸附结构如图5(b)所示。根据这种吸附结构,可以得到:
HCHO+H2O→CH2+2OH
并且仅在600 K下观察到此现象,这导致了在此温度下的吸附能是最低的。结果表明,湿度在α-Fe2O3(100)表面与甲醛之间的化学反应中起着重要的作用。
4 结论
使用ReaxFF可以计算大量分子的体系,在一些方面可以获得与量子力学相同的结果,计算时间显著缩短,明显提升计算效率,适合用于气体在物质的表面吸附的仿真分析。
对氧化铁(100)表面气体吸附现象的ReaxFF仿真计算结果可知,不同于SnO2、ZnO等n型材料,氧气对甲醛在氧化铁表面上的吸附没有明显影响,甲醛分子直接吸附在氧化铁(100)表面上。在4种气体中,甲醛最容易吸附在氧化铁表面上,其次是水,然后是氢气,最后是氧气,并且高温下氧气几乎不会吸附。在甲醛在氧化铁(100)表面上的吸附中湿度起着重要的作用,水对于甲醛的吸附和分解有促进作用。
猜你喜欢
中国音乐学(2022年1期)2022-05-05
粉末冶金技术(2021年1期)2021-03-29
生物学通报(2021年4期)2021-03-16
江苏安全生产(2020年1期)2020-03-16
物理学报(2018年10期)2018-06-14
无机盐工业(2017年5期)2017-03-11
上海金属(2016年4期)2016-11-23
上海金属(2016年3期)2016-11-23
中央民族大学学报(自然科学版)(2014年3期)2014-06-09
中国质量与标准导报(2014年6期)2014-02-28
