
稳定及提高WO3−x光催化活性的研究进展
2021-04-05 12:43赵冬雪王芳芳
石油化工高等学校学报 2021年1期
赵冬雪,张 静,王芳芳
(辽宁石油化工大学辽宁省先进清洁能源材料化学工程实验室,辽宁抚顺113001)
随着世界经济的快速发展,能源短缺问题越来越严重,环境污染问题也越来越突出。A.Fujishima等[1]发现TiO2在光电催化条件下,可将水分解生成氢气;B.Kraeutler等[2]发现TiO2半导体材料可以通过光催化技术降解水中污染物。光催化技术具有反应条件温和、无二次污染以及成本低等优点[3],因此利用光催化技术解决能源危机和环境问题引起了国内外研究者的广泛关注。
一般来说,基于半导体材料的光催化反应分为三个步骤[4]:一是在光激发下,半导体吸收大于或等于其禁带宽度的能量,价带上的电子被激发跃迁到导带,同时在价带上留下空穴,从而产生光生电子⁃空穴对;二是光生电子⁃空穴对分离并分别迁移到半导体表面,在迁移过程中,电子和空穴还会在半导体的体内或表面发生复合;三是迁移到半导体表面的空穴和电子分别与吸附在半导体表面的反应物发生氧化和还原反应。根据光催化反应的三个步骤,影响催化剂光催化性能的主要因素有半导体的能带位置、光生电子⁃空穴对的分离效率、比表面积以及半导体的晶粒尺寸等[5]。
目前大多数催化剂由于其电子⁃空穴复合率高或光的利用率低等因素,限制了它们在光催化领域的实际应用。国内外研究者通常采用离子掺杂、形貌调控、构建“结”结构、构建缺陷等策略来提高光催化效率。离子掺杂是通过掺杂金属离子或非金属离子调节半导体禁带宽度使其变窄或产生中间能级,导致半导体的吸光范围扩展到可见光,从而提高对可见光的利用率[6⁃8];通过形貌调控合成多级结构等特殊形貌,可增大光催化剂的比表面积,增强光的吸收,提高电子⁃空穴对的分离效率,进而提高光催化活性[9⁃12];“结”结构的构建是通过在两种半导体的界面形成内建电场而促进电子⁃空穴对的分离[13⁃14],此外不同禁带宽度的半导体材料构成“结”结构后,还可以增强光吸收[15⁃16],进而提高光催化活性;在半导体中构建缺陷,可以改变能带结构,增强光吸收,还可以通过电子⁃空穴对分离以及提高迁移速率来提高催化剂的光催化活性[17⁃19]。目前,在半导体中构建缺陷已成为众多光催化剂改性策略中的重要方法之一。
WO3是一种在可见光范围内具有光响应的半导体材料,具有抗光腐蚀、廉价和无毒等优点,所以基于WO3的光催化技术具有广泛的应用前景,但是WO3仍然存在光生电子⁃空穴对的复合效率高、电子转移速率低、导带位置低等缺点[20],使WO3的光催化活性较低。研究结果表明[21],通过在WO3中引入氧空位,形成非化学计量比的氧化钨(WO3−x)可以提高其光催化性能。但O2或H2O倾向于吸附在WO3−x的 氧 空 位,使WO3−x易 被 氧 化[22]。WO3引 入氧空位后不稳定,所以研究如何稳定WO3−x和进一步提高其光催化性能具有重要的意义。本文基于以上研究结果,对该问题进行梳理总结,首先探讨了氧空位影响WO3催化剂活性的机理,然后讨论了如何稳定WO3−x和进一步提高其光催化活性,并对WO3−x在未来光催化领域应用面临的挑战和机遇进行了展望。
1 氧空位对光催化性能的影响机制
多数的光催化材料都存在晶体缺陷,普遍认为缺陷是光生电子⁃空穴对的复合中心和散射中心,由于两者均不利于载流子的迁移,从而使光催化活性降低[23]。随着人们的深入研究,逐渐发现缺陷对提高光催化性能也具有积极作用[24],因此国内外研究者进一步研究了缺陷对催化剂光催化活性的影响。
根据晶格维数,氧化物中的缺陷通常分为点缺陷、线缺陷和面缺陷[25⁃27]。非化学计量氧化物中的点缺陷主要包括空位、取代(置换)式和间隙式。空位是正常结点没有被原子或离子占据所形成的空节点;外来原子进入晶格成为晶体中的杂质,称为取代(置换)式;原子进入晶体正常结点之间的间隙位置,称为间隙式。空位是光催化半导体材料中普遍存在的点缺陷,尤其是氧空位已经被广泛报道存在于很多金属氧化物半导体光催化剂中,如等 氧 化 物。研 究 表 明[31⁃32],当WO3中引入氧空位后,光催化活性提高,主要原因是氧空位改变了它的能带结构,从而增强了WO3对光的吸收,抑制了光生电子⁃空穴对的复合,提高了载流子的迁移速率并改变其导电性。
1.1 对能带结构和吸光性质的影响
根据分子轨道理论,金属氧化物半导体的导带是由金属原子的d轨道提供,价带则是由O原子的2p轨道提供[33]。当金属氧化物引入氧空位后,氧空位周围的原子将引起化学环境的变化,导致电子的再分配[27],进而造成氧化物的能带结构的变化。N.Zhang等[34]发现含有丰富氧空位的WO3(R⁃WO3)与含少量氧空位的WO3(D⁃WO3)相比,其价带顶上移,导带底下移,禁带宽度变窄。由此可知,当WO3中引入缺陷后,可以使其禁带宽度变窄。N.Serpone等[35]认为样品吸收带边红移的原因是显色中心的形成,即样品的颜色发生变化。当WO3中引入氧空位后,其样品颜色会随着氧空位含量的增多而不断加深,因此其吸光度向可见光/近红外范围延伸。如Q.Liu等[36]通过在真空条件下焙烧WO3,制备了具有氧空位含量的WO3−x。随着焙烧温度不断升高,样品颜色由黄色变为浅绿色,最终变为深蓝色,样品的吸收带边缘也逐渐向长波方向移动,并且依然保持对紫外光的吸收。
近几年研究发现,当WO3−x受到光激发后,晶体中所有自由电荷载流子形成集体振荡,从而引起局域表面等离子共振(LSPR)效应。强烈的LSPR效应会增 强光吸收[37⁃38]。
1.2 对电子⁃空穴对分离效率的影响
适量的氧空位可作为光生电子的捕获中心,从而抑制光生电子⁃空穴对的复合而提高光催化活性,而过量的氧空位则成为电子⁃空穴对的复合中心,使电子⁃空穴对的分离速率降低,进而降低光催化活性[39⁃40]。J.Meng等[41]通 过 冷 却 的 方 法 控 制 氧 空 位 浓度,当适量氧空位引入WO3时,可以抑制电子⁃空穴对的复合,进而使WO3−x的光催化活性得到提高。Y.Li等[42]研究结果表明,当WO3中引入一定量的氧空位之后,可以俘获和转移电子,从而降低电子⁃空穴对的复合效率。X.Zhou等[43]通过氢化法制备了表面含有氧空位的WO3,由于氧空位的引入使WO3−x的带隙变窄,增强WO3−x的可见光吸收,使电荷的转移速率增强。
1.3 对导电性的影响
氧空位的引入会导致氧化物的导电性发生变化[44]。例 如,非 化 学 计 量 比 的WO3−x(如W18O49、WO2.8、WO2.83和WO2.9等),随着氧空位数量的变化,其导电性质也会发生变化[45]。当0<x<0.1时,电学性质主要由极化子中局部电子支配,因此WO3−x表现出半导体性质;当x=0.1时,由于局部极化子开始重叠并且形成离域态,WO3−x发生金属⁃绝缘体转变(Mott转变);当x>0.1时,电化学性质主要由自由电子主导,WO3−x呈现出金属的导电性。
2 WO3−x改性策略
WO3−x在溶液中容易被氧化,导致WO3−x的稳定性相对较差[23],为了增强WO3−x的稳定性,通常采用稳定LSPR效应的方法[46]。同时,有研究采用贵金属或含碳材料与氧空位协同方法提高光催化活性。
2.1 稳定WO3−x的方法
WO3−x在溶液中被氧化后,使WO3−x中氧空位含量减少,从而导致自由电子数量减少,减弱LSPR效应,光催化活性不稳定。因此,为了稳定WO3−x光催化剂,必须不断地向WO3−x中迁移电子。Z.Lou等[47]为 了 解 决WO3−x在 水溶 液 中 不稳 定 的 问题,首先设计合成了CdS/WO3−x异质结构,发现CdS受光激发后,价带的电子被激发跃迁到导带,然后连续注入WO3−x导带,使WO3−x导带中自由电子的数量增加(见图1),从而维持了WO3−x的LSPR效应。因此,在可见光/近红外光照射下,CdS/WO3−x具有较高的稳定性,光解水活性也比WO3−x和CdS高。该实验证明,通过构建异质结可使电子连续不断地迁移到WO3−x的导带,稳定其LSPR效应,进而实现稳定WO3−x的目的。
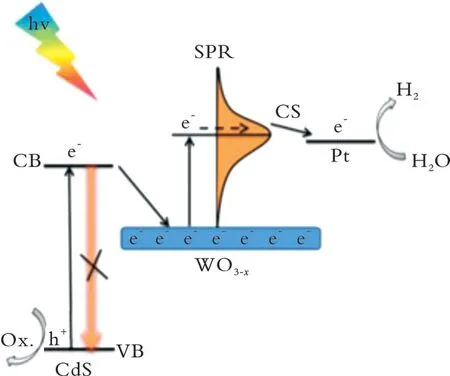
图1 半导体CdS向WO3−x注入光生电子示意Fig.1 Schematic diagram of semiconductor CdS injection into WO3-x
为了进一步研究如何稳定WO3−x,该课题组又通过溶剂热法将介孔TiO2(TiO2⁃MCs)与WO3−x纳米线(WO3−x⁃NWs)耦合,制备了异质结构TiO2⁃MCs/WO3−x⁃NWs[48]。结果表明,TiO2受光激发后,价带上产生的电子被激发跃迁到导带,导带上的光电子发生转移(CT1、CT2和CT3),TiO2导带上发生转移的光电子到达TiO2表面缺陷能级(ES)并且不断注入WO3−x的导带(CB)和导带的最低空能级(EV)(见图2)。由于光电子的连续注入稳定了WO3−x导带上的载流子浓度,进而稳定了WO3−x的LSPR效应,使TiO2⁃MCs/WO3−x⁃NWs在光催化还原CO2时,表现出很好的稳定性,而且其活性比TiO2⁃MCs和WO3−x⁃NWs都要高。实验结 果证明,稳定WO3−x最重要的方法是向其导带不断转移电子。
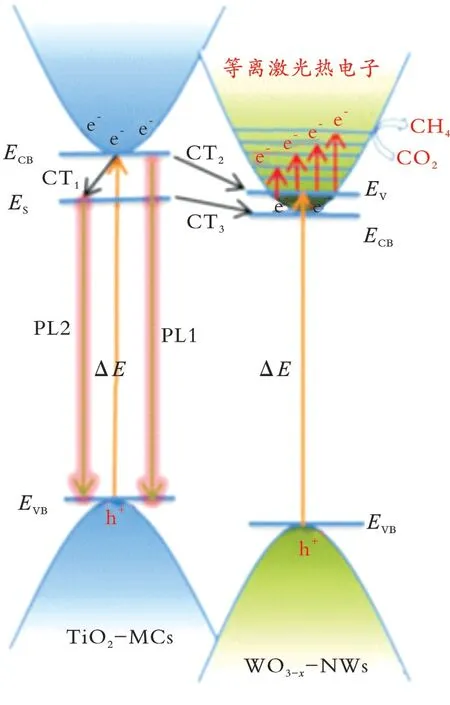
图2 TiO2⁃MCs/WO3−x⁃NWs中光电子的产生和转移Fig.2 Generation and transfer of photoelectrons in TiO2⁃MCs/WO3-x⁃NWs
2.2 提高WO3−x光催化活性的方法
2.2.1 WO3−x与贵金属的协同作用 WO3−x在可见光/近红外照射下,表现出强烈的LSPR效应,而贵金属纳米粒子Ag或Au也具有极强的LSPR效应。当WO3−x与贵金属复合时,在近红外/可见光的照射下,WO3−x和贵金属LSPR效应的协同作用可以促进光生电子⁃空穴对的分离,并提高电子的迁移速率,进而可以提高WO3−x的光催化活性[49]。
J.Chen等[50]设计合成了Ag/WO3−x复合光催化材料,在近红外光照下,WO3−x产生高能的电子,这些电子很快地转移到Ag上,导致Ag表面载流子密度增加,从而使Ag表面带负电,WO3−x带正电,有利于电荷的快速迁移,提高Ag/WO3−x复合光催化材料的光催化活性。K.Chen等[51]采用溶剂热法合成了Au@WO3−x复合材料。该复合材料在可见光和近红外照射下,Au核产生的电子转移到WO3−x壳上,WO3−x充当催化位点,有助于电子⁃空穴对的分离和转移,从而使其光催化活性提高。综上研究表明,利用WO3−x与贵金属的LSPR效应的协同作用可作为一种有效提高WO3−x光催化活性的方法。
2.2.2 WO3−x与含碳材料的协同作用 当含碳材料与WO3−x形成复合材料时,含碳材料会增大WO3−x的比表面积,进而增加与反应物接触的几率,同时增加复合材料的导电性,提高复合材料的光催化活性[52]。
P.Chen等[53]采用超快速溶液燃烧合成方法,制备了非晶碳包裹WO3−x纳米材料。由于非晶碳的存在,使催化剂对有机分子的吸附增强,另外碳可以抑制WO3−x晶粒的生长,在燃烧过程中碳的存在使WO3向WO3−x和W18O49转 变,使 氧 空 位 引 入WO3中,有利于增强WO3−x对可见光的吸收,而且氧空位为光生电子提供捕获位点,促进了电子的转移,因此复合材料的光催化活性得到显著提高。L.Wang等[54]合成了WO3−x和三维氮掺杂碳组成的复合光催化剂(WO3−x/NC)。三维氮掺杂碳增加了WO3−x与甲醛的接触,而且WO3−x和三维氮掺杂碳导电性的协同作用,提高了WO3−x的电子输运性能,使复合材料具有良好的去除挥发性有机物(VOCs)的性能。这些研究表明,提高WO3−x光催化活性可通过提高其比表面积以及增加其导电性来实现。
2.2.3 其他方法 M.Guo等[55]通过两步水热法制备了WS2/MoS2@WO3−xZ型核壳异质结构,该催化剂在光降解2,4⁃二氯苯酚时,表现出良好的光催化活性,这归因于MoS2堆积形成了具有较多边缘活性位和较大比表面积的纳米球;氧空位和异质结的形成有利于光生电子⁃空穴对的空间分离和转移,使光生电子⁃空穴对更容易在催化剂表面聚集并参与反应;催化剂的光吸收范围增大,提高了光的吸收和利用率。Y.Bu等[56]采用原位沉积法制备了Z型Ag3PO4/Ag/WO3−x异质结,其中WO3−x在可见光照射下产生的光生电子转移到Ag上,Ag3PO4在光照下产生的空穴也向Ag转移,从而导致还原性弱的光生电子和氧化性弱的光生空穴湮灭,有效地延长了Ag3PO4产生的光电子和WO3−x中空穴的寿命,提高了其光催化降解罗丹明B、甲基橙等有机染料的性能。B.Li等[57]利用两步原位负载过程构建了Ag/WO2.72/还原氧化石墨烯(rGO)或Au/WO2.72/rGO三元纳米复合光催化剂,其中WO2.72纳米线具有可见光催化活性,且对反应底物表现出很高的吸附特性,金属纳米粒子进一步扩宽了三元纳米复合催化剂的可见光响应范围,同时提高了光生载流子的寿命,rGO则为光生电子的快速转移提供了通道。因此,该三元纳米复合光催化剂比单一WO2.72纳米线具有更高的光催化活性。以上研究表明,增加WO3−x的比表面积,促进其电子⁃空穴的分离和转移以及增强其吸光性是提高WO3−x光催化活性的有效方法。
3 结论与展望
WO3作为一种常用催化剂在光催化领域深受欢迎。当WO3中引入氧空位后,使禁带宽度发生改变,吸光范围延伸至可见/近红外区域,同时适量氧空位的引入还会促进电子⁃空穴的分离和迁移速率,增强其光催化活性,所以WO3−x是一种应用前景广阔的光催化材料。但由于WO3−x不稳定,阻碍了WO3−x在光催化领域的应用。因此,未来对WO3−x的研究仍需要进一步探索,以期提高WO3−x稳定性及光催化活性。同时,研究探寻高效环保节能复合方法,降低资源损耗及成本,以实现拓宽WO3−x的应用范围。相信随着WO3−x催化剂研究的不断深入,未来WO3−x催化剂在光催化领域将有更加广阔的应用和发展前景。
猜你喜欢
科学之友(2022年11期)2022-11-03
物理学报(2022年17期)2022-09-14
辽宁石油化工大学学报(2021年6期)2022-01-04
装备维修技术(2021年36期)2021-10-25
弹箭与制导学报(2021年3期)2021-07-30
石油化工高等学校学报(2021年3期)2021-07-15
无机盐工业(2020年1期)2020-12-31
网印工业(2019年4期)2019-05-21
作文中学版(2018年1期)2018-11-28
读者欣赏(2014年6期)2014-07-03
