
GSK-A1及其衍生物的合成
2021-04-02 02:11周志旭赵春深
合成化学 2021年3期
任 倩,刘 叶,朱 雄,周志旭,赵春深*
(1.贵州大学 药学院,贵州 贵阳 550025;2.贵州省合成药物工程实验室,贵州 贵阳 550025)
丙型肝炎是由丙型肝炎病毒(HCV)导致的一种与乙型肝炎相似的病毒性肝炎,患者多为慢性携带者,HCV蛋白多功能的发现给抗HCV药物的研究带来了灵感。目前已知的药物靶标有非结构蛋白NS5A和HCV酶NS3/4A、NS5B[1]。NS5A是一种高度磷酸化的非结构蛋白,不具备酶催化活性,其磷酸化水平在HCV基因组的复制和翻译过程中起调节的作用。脂酰肌醇4-激酶IIIα(PI4KIIIα)与NS5A作用刺激产生脂质磷脂酰肌醇4-磷酸(PI4P)的脂质激酶[2-3],PI4P可参与病原性RNA病毒的复制。PI4P是产生信号传导脂质PI(4,5)P2和PI(3,4,5)P3的前体,在调节病原体病毒感染中发挥关键作用[4]。在HCV感染过程中,PI4P水平上升,5-(2-氨基-1-(4-吗啉代苯基)-1H-苯并[d]咪唑-6-基)-N-(2-氟苯基)-2-甲氧基吡啶-3-磺酰胺(GSK-A1)可通过抑制PI4KIIIα干扰PI4P的供应进而抑制HCV的复制[5-6]。
2012年,Botyanszki J和Dickerson S H等合成了一系列化合物,中间涉及GSK-A1的合成[7-8],这也是目前报道的唯一合成路线。但是该合成路线不适合中试放大生产,如使用的原料5-溴-2-氯-3-氨基吡啶、4-溴-2-氟硝基苯价格昂贵;多步反应的后处理过程较为复杂,需采用柱层析纯化,效率和产率都较低。本文以反应可行性、原料廉价易得、反应操作简单、反应条件温和、后处理简洁及收率高的原则出发,设计了一条GSK-A1的合成路线。以5-溴-2-甲氧基吡啶、吗啉、2-氟苯胺和联硼酸频那醇酯等为原料,经过硝基化[9-10]、磺酰化,硼酸酯化[11]和Suzuki-Miyaura[12-15]等反应合成目标GSK-A1(Scheme 1),总收率19.33 %,并在此基础上设计合成了9个新的衍生物(Scheme 2),其结构经1H NMR和HR-MS(ESI-TOF)确证。且在实验的过程中得到了中间体5-溴-2-甲氧基-3-磺酰氯吡啶,为后续GSK-A1衍合物的合成提供了思路。

Scheme 1
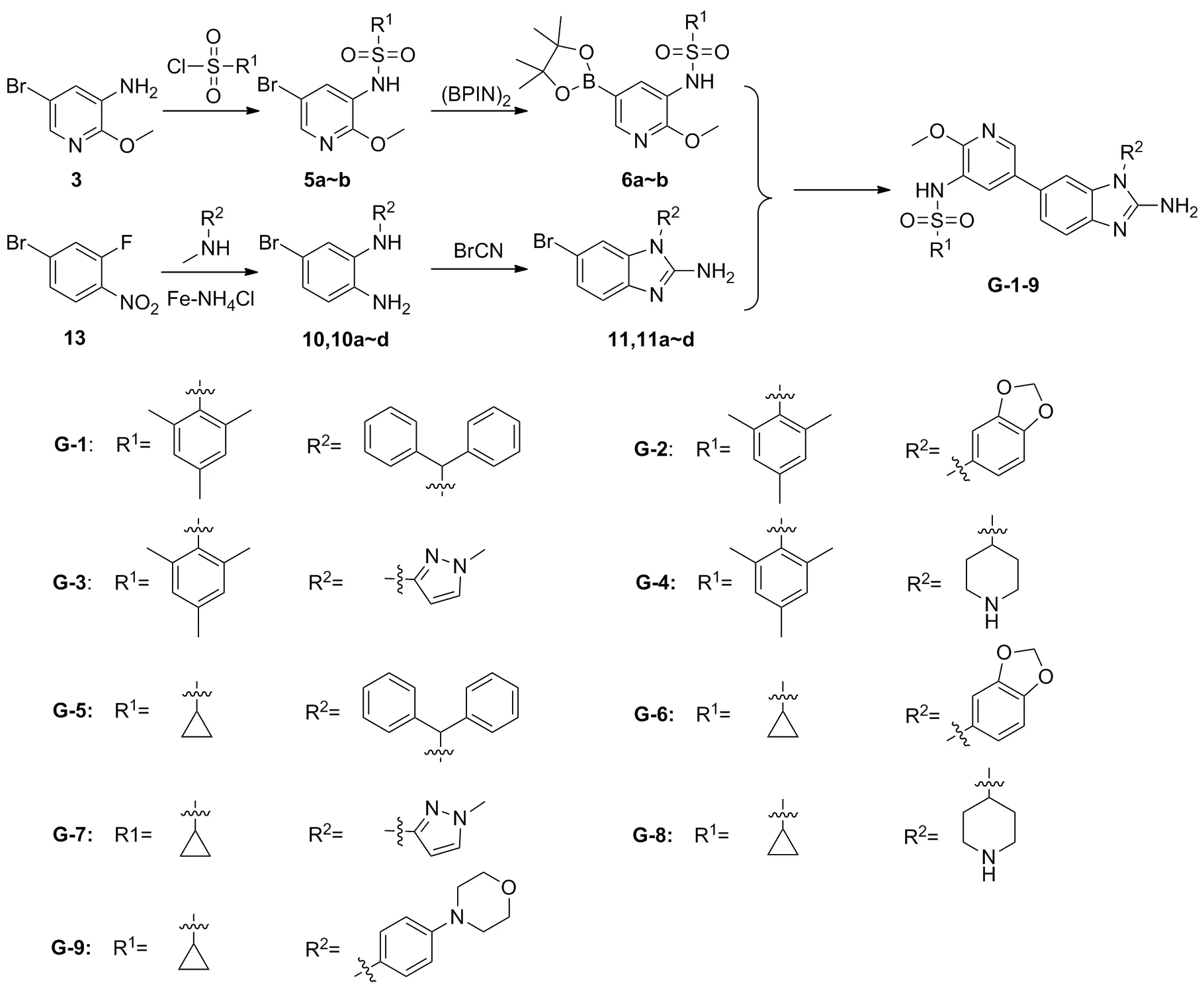
Scheme 2
1 实验部分
1.1 仪器与试剂
T-4型熔点仪;Advance DMX 400 MHz型核磁共振仪(THR-MS为内标);Agilent 1100 LC/HR-MS型质谱仪。
所用试剂均为化学纯或分析纯。
1.2 合成
(1) 5-溴-2-甲氧基-3-硝基吡啶(2)的合成
冰浴下,在500 mL三口瓶中加入100 mL浓硫酸和5-溴-2-甲氧基吡啶30 g(159.56 mmol),控温0~5 ℃,缓慢滴加100 mL浓硝酸,滴完搅拌1 h后升至室温搅拌5.5 h(TLC检测)。反应完毕,将反应液慢慢倒入装有300 mL冰块的烧杯中,析出固体,抽滤,滤饼用清水(3×100 mL)洗涤,干燥后得黄色固体230.7 g,收率82.5%,m.p.88~92 ℃;1H NMR(CDCl3,400 MHz)δ:8.45(d,J=2.3 Hz,1H),8.39(d,J=2.3 Hz,1H),4.11(s,3H)。
(2) 3-氨基-5-溴-2-甲氧基吡啶(3)的合成
将230 g(128.74 mmol)溶于300 mL乙醇中,加入饱和NH4Cl水溶液20.7 g(386.23 mmol),冰浴下加入Fe粉35.9 g(643.72 mmol),室温搅拌3.5 h(TLC检测)。反应完毕,硅藻土辅助过滤除去铁泥,滤液用乙酸乙酯(3×200 mL)萃取,合并有机相,无水硫酸钠干燥,减压蒸除溶剂得棕色固体324.4 g,收率93.4%,m.p.60~62 ℃;1H NMR(400 MHz,CDCl3)δ:7.58(d,J=2.1 Hz,1H),6.97(d,J=2.1 Hz,1H),3.95(s,3H),3.85(s,2H),粗品不经处理直接用于下一步反应。
(3) 5-溴-2-甲氧基-3-磺酰氯吡啶(4)的合成
称取SOCl2(50 mL)滴加至冰水(360 mL)中,控温0 ℃,搅拌17 h后,加入氯化亚铜146.3 mg(1.48 mmol);0 ℃下,在500 mL三口瓶中加入浓盐酸(190 mL)和325g(123.13 mmol),滴加60 mL NaNO2溶液(含NaNO210.2 g,147.75 mmol),滴毕,-5~0 ℃下搅拌15 min。然后将该溶液滴加至氯化亚铜的溶液中,0 ℃下搅拌80 min(TLC检测)。反应完毕,抽滤,滤饼用清水(3×200 mL)洗涤,干燥得黄色固体428.4 g,收率80.6%,m.p.91~94 ℃;1H NMR(CDCl3,400 MHz)δ:8.52(d,J=2.4 Hz,1H),8.33(d,J=2.4 Hz,1H),4.18(s,3H)。
(4) 5-溴-N-(2-氟苯基)-2-甲氧基-3-磺酰胺吡啶(5)的合成
在500 mL三口瓶中,依次加入邻氟苯胺9.3 g(83.76 mmol)、420 g(69.8 mmol)、吡啶11 g(139.6 mmol)和150 mL二氯甲烷,室温搅拌5 h(TLC检测)。反应完毕,减压浓缩除去溶剂,加200 mL清水析出固体,抽滤,滤饼用清水(3×100 mL)洗涤,干燥后得浅褐色固体523.2 g,收率92.2%,m.p.152~155 ℃;1H NMR(CDCl3,400 MHz)δ:8.32(d,J=2.3 Hz,1H),8.15(d,J=2.3 Hz,1H),7.57~7.51(m,1H),7.30(s,1H),7.07(d,J=4.8 Hz,2H),6.98(t,J=5.4 Hz,1H),4.10(s,3H)。
(5)N-(2-氟苯基)-2-甲氧基-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)-3-磺酰胺吡啶(6)的合成
将517 g(47.07 mmol)溶于250 mL 1,4-二氧六环中,依次加入联硼酸频那醇酯12 g(47.07 mmol),乙酸钾13.8 g(141.20 mmol)和Pd(dppf)2Cl2172.2 mg(0.24 mmol),氮气保护下升温至100 ℃反应4 h(TLC检测)。反应完毕,将反应液倒入200 mL清水中,乙酸乙酯(3× 200 mL)萃取,合并有机相,无水硫酸钠干燥,减压蒸除溶剂,残余经硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=5/1,V/V)纯化得白色固体615.7 g,收率81.8%,m.p.177~180 ℃;1H NMR(CDCl3,400 MHz)δ:8.62(d,J=1.8 Hz,1H),8.44(d,J=1.8 Hz,1H),7.59(dd,J=7.6 Hz,2.5 Hz,1H),7.29(d,J=2.8 Hz,1H),7.05(dd,J=6.4 Hz,3.1 Hz,2H),7.00~ 6.94(m,1H),4.14(s,3H),1.31(s,12H)。
(6) 4-(4-硝基苯基)吗啉(8)的合成
将吗啉18 g(206.6 mmol)溶于乙腈(150 mL)中,加入729.2 g(206.6 mmol)和碳酸钾57.1 g(413.22 mmol),加热至回流,反应4 h(TLC检测)。反应完毕,将反应液倒入200 mL清水中,析出固体,抽滤,滤饼用清水(3×100 mL)洗涤,干燥得黄色固体842.2 g,收率98.3%,m.p.147~150 ℃;1H NMR(CDCl3,400 MHz)δ:8.15(s,1H),8.13(s,1H),6.85(s,1H),6.82(s,1H),3.88~3.85(m,2H),3.39~3.36(m,2H)。
(7) 4-吗啉代苯胺(9)的合成
将820 g(96.06 mmol)溶于乙醇(200 mL)中,加入饱和NH4Cl水溶液15.4 g(288.18 mmol),冰浴下加入铁粉26.8 g(480.31 mmol)反应4.5 h(TLC检测)。反应完毕,硅藻土辅助过滤除去铁泥,滤液用乙酸乙酯(3×200 mL)萃取,合并有机相,无水硫酸钠干燥,减压蒸出溶剂得深灰色固体915.8 g,收率92.7%,m.p.120~123 ℃;1H NMR(CDCl3,400 MHz)δ:8.17~8.11(m,2H),6.86~6.80(m,2H),3.89~3.85(m,4H),3.39~3.36(m,4H),粗品不经处理直接用于下一步反应。
(8) 5-溴-N-(4-吗啉代苯基)苯-1,2-二胺(10)的合成
将912.2 g(68.45 mmol)溶于乙腈(150 mL)中,加入4-溴-2-氟硝基苯15 g(68.45 mmol)和碳酸钾18.8 g(136.9 mmol),加热至78 ℃回流反应5 h(TLC检测)。反应完毕,将反应液倒入200 mL清水中,析出固体,抽滤,滤饼用清水(3×100 mL)洗涤,干燥得橙色固体5-溴-N-(4-吗啉代苯基)-2-硝基苯胺25.6 g,收率97.9%。
将5-溴-N-(4-吗啉代苯基)-2-硝基苯胺20 g(52.88 mmol)溶于200 mL 乙醇中,加入饱和的NH4Cl水溶液8.5 g(158.63 mmol),冰浴下加入Fe粉14.8 g(264.39 mmol)反应4 h(TLC检测)。反应完毕,硅藻土辅助过滤除去铁泥,滤液用乙酸乙酯(3×200 mL)萃取,合并有机相,无水硫酸钠干燥,减压蒸出溶剂得深灰色固体1017.8 g,收率92%,m.p.125~128 ℃;1H NMR(CDCl3,400 MHz)δ:7.12(s,1H),6.98(d,J=6.4 Hz,1H),6.90~6.77(m,4H),6.64(d,J=8.3 Hz,1H),5.04(s,1H),3.88~3.84(m,4H),3.64(s,2H),3.08(s,4H),粗品不经处理直接用于下一步反应。
(9) 6-溴-1-(4-吗啉代苯基)-1H-苯并[d]咪唑-2-胺(11)的合成
将1011 g(31.59 mmol)溶于MeOH/H2O(50 mL/50 mL)中,加入氰化溴5.4 g(50.54 mmol),50 ℃回流反应2 h(TLC检测)。反应完毕,减压除去溶剂甲醇,残余物倒入少量50 mL清水中,析出固体,抽滤,滤饼用碳酸氢钠水溶液(3×100 mL)洗涤,抽滤干燥后即得白色固体119.8 g,收率83.4%,m.p.228~231 ℃;1H NMR(CDCl3,400 MHz)δ:7.45(s,1H),7.41(d,1H),7.32(d,J=8.9 Hz,3H),7.16(s,1H),7.11(d,J=9.0 Hz,2H),3.92~3.89(m,4H),3.33~3.30(m,4H)。
(10) 5-(2-氨基-1-(4-吗啉代苯基)-1H-苯并[d]咪唑-6-基)-N-(2-氟苯基)-2-甲氧基吡啶-3-磺酰胺(12)的合成
在1,4-二氧六环/水(120 mL/10 mL)混合溶剂中,依次加入62 g(4.9 mmol),111.83 g(4.9 mmol),Pd(PPh3)40.17 g(0.15 mmol)和碳酸钾2 g(14.7 mmol),氮气保护,升温至100 ℃反应16 h(TLC检测)。反应完毕,将反应液倒入200 mL清水中,乙酸乙酯(3× 200 mL)萃取,合并有机相,无水硫酸钠干燥,减压蒸出溶剂,甲基叔丁基醚(2×10 mL)洗涤,抽滤后得白色固体 5-(2-氨基-1-(4-吗啉代苯基)-1H-苯并[d]咪唑-6-基)-N-(2-氟苯基)-2-甲氧基吡啶-3-磺酰胺121.69 g,收率60.3%,m.p.252~255 ℃;1H NMR(DMSO-d6,400 MHz)δ:8.67(s,1H),8.13(s,1H),7.30(d,J=8.5 Hz,3H),7.15(dd,J=6.1 Hz,2.6 Hz,7H),6.83(d,J=1.2 Hz,1H),3.95(s,3H),3.80~3.76(m,4H),3.24~3.20(m,4H);HR-MS(ESI-TOF)m/z:Calcd for C29H27N6O4FS{[M+H]+}575.1860,found 575.1871。
1.3 GSK-A1衍生物及其中间体的合成
(1) 衍生物中间体(6a~b)的合成
将311.1 g(54.87 mmol)和吡啶7.2 g(91.45 mmol)溶于150 mL DCM中,加入2,4,6-三甲基苯磺酰氯10 g(45.72 mmol),室温搅拌5 h(TLC检测)。反应完毕,减压浓缩除去溶剂,残余物倒入少量50 mL清水中,析出固体,抽滤,滤饼用清水(3×100 mL)洗涤,干燥后得5a。
将5a10 g(25.95 mmol)溶于150 mL 1,4-二氧六环中,加入联硼酸频那醇酯6.6 g(25.95 mmol),乙酸钾7.6 g(77.87 mmol)和Pd(dppf)2Cl294.96 mg(0.13 mmol),氮气保护下升温至100 ℃反应4 h(TLC检测)。反应完毕,将反应液倒入200 mL清水中,乙酸乙酯(3×100 mL)萃取,合并有机相,无水硫酸钠干燥,减压蒸除溶剂,残余经硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=5/1,V/V)纯化得6a。将原料换为环丙烷磺酰氯,同法合成得6b。
(2) 衍生物中间体(11a~11d)的合成
将二苯甲胺10 g(54.57 mmol)溶于150 mL乙腈中,加入4-溴-2-氟硝基苯12 g(54.57 mmol)和碳酸钾15.1 g(109.14 mmol),升温至78 ℃反应5 h(TLC检测)。反应完毕,将反应液倒入200 mL清水中,有固体析出,抽滤,滤饼用清水(3×100 mL)洗涤,干燥得N-二苯甲基-5-溴-2-硝基苯胺。
将N-二苯甲基-5-溴-2-硝基苯胺15 g(39.14 mmol)溶于200 mL乙醇中,加入饱和的NH4Cl水溶液6.3 g(117.42 mmol),冰浴下加入Fe粉10.9 g(195.69 mmol),反应4 h(TLC检测)。反应完毕,硅藻土辅助过滤除去铁泥,滤液用乙酸乙酯(3×100 mL)萃取,合并有机相,用无水硫酸钠干燥,减压蒸除溶剂得10a。
将10a10 g(28.31 mmol)溶于MeOH/H2O(50 mL/50 mL)混合溶剂中,加入BrCN 4.8 g(45.29 mmol),50 ℃回流反应2 h(TLC检测)。反应完毕,减压除去溶剂甲醇,残余物倒入少量50 mL清水中,析出固体,抽滤,滤饼用碳酸氢钠水溶液(3×100 mL)洗涤,抽滤干燥得11a。将原料分别换为4-氨基-N-Boc哌啶,3,4-亚甲二氧基苯胺和N-甲基-3-氨基吡唑,同法合成得11b~11d。
(3)GSK-A1衍生物(G-1~G-9)的合成
在1,4-二氧六环/水(120 mL/10 mL)混合溶剂中,加入6a2 g(4.63 mmol),11a1.75 g(4.63 mmol),Pd(PPh3)40.16 g(0.14 mmol)和碳酸钾1.9 g(13.88 mmol),氮气保护,升温至100 ℃反应16 h(TLC检测)。反应完毕,将反应液倒入200 mL 清水中,乙酸乙酯(3×100 mL)萃取,合并有机相,无水硫酸钠干燥,减压蒸除溶剂得到粗品,用甲基叔丁基醚洗涤,抽滤即得白色固体N-(5-(2-氨基-1-二苯甲基-1H-苯并[d]咪唑-6-基)-2-甲氧基吡啶-3-基)-2,4,6-三甲基苯磺酰胺(G-1) 1.75 g,收率61.4%;1H NMR(DMSO-d6,400 MHz)δ:7.46~7.34(m,10H),7.19(t,J=7.1 Hz,7H),7.08(s,1H),6.92(s,2H),6.43(s,1H),3.58(s,3H),2.40(s,6H),2.20(s,3H);HR-MS(ESI-TOF)m/z:Calcd for C35H33N5O3S{[M+H]+}604.2357,found 604.2377。
同法,中间体6a~6b分别与11,11a~11d反应合成得到衍生物G-2~G-9。
N-(5-(2-氨基-1-(苯并[d] [1,3]二恶唑-5-基)-1H-苯并[d]咪唑-6-基)-2-甲氧基吡啶-3-基)-2,4,6-三甲基苯磺酰胺(G-2):灰白色固体1.51 g,收率57.2%;1H NMR(DMSO-d6,400 MHz)δ:7.26(d,J=8.2 Hz,1H),7.17(dd,J=11.7 Hz,7.5 Hz,3H),7.10(d,J=2.0 Hz,1H),6.95(d,J=8.1 Hz,2H),6.86(s,2H),6.75(s,1H),6.20(s,2H),3.68(s,3H),2.45(s,6H),2.19(s,3H);HR-MS(ESI-TOF)m/z:Calcd for C29H27N5O5S{[M+H]+}558.1795,found 558.1806。
N-(5-(2-氨基-1-(1-甲基-1H-吡唑-3-基)-1H-苯并[d]咪唑-6-基)-2-甲氧基吡啶-3-基)-2,4,6-三甲基苯磺酰胺(G-3):白色固体1.42 g,收率57.7%;1H NMR(DMSO-d6,400 MHz)δ:8.08(s,1H),7.99(s,1H),7.54(s,2H),7.30(d,J=7.8 Hz,1H),7.24~7.16(m,3H),6.94(s,3H),6.59(s,1H),3.96(s,3H),3.68(s,3H),2.49(s,6H),2.20(s,3H);HR-MS(ESI-TOF)m/z:Calcd for C26H27N7O3S{[M+H]+}518.1953,found 518.1969。
N-(5-(2-氨基-1-(哌啶-4-基)-1H-苯并[d]咪唑-6-基)-2-甲氧基吡啶-3-基)-2,4,6-三甲基苯磺酰胺(G-4):白色固体1.68 g,收率66.1%;1H NMR(DMSO-d6,400 MHz)δ:8.10(s,1H),7.62(s,1H),7.38(s,1H),7.21(d,J=8.1 Hz,2H),7.02(d,J=8.6 Hz,2H),6.97(s,3H),4.35(s,1H),3.64(s,6H),3.24(t,J=13.1 Hz,4H),2.21(s,6H),1.80(d,J=13.3 Hz,4H);HR-MS(ESI-TOF)m/z:Calcd for C27H32N6O3S{[M+H]+}521.2290,found 521.2390。
N-(5-(2-氨基-1-二苯甲基-1H-苯并[d]咪唑-6-基)-2-甲氧基吡啶-3-基)环丙烷磺酰胺(G-5):白色固体1.61 g,收率52.8%;1H NMR(DMSO-d6,400 MHz)δ:7.78(s,1H),7.49(d,J=2.2 Hz,1H),7.41(dt,J=12.7 Hz,6.8 Hz,9H),7.23(s,1H),7.20(d,J=7.2 Hz,5H),7.09(s,1H),6.44(s,1H),3.89(s,3H),1.23(s,1H),0.91(d,J=7.5 Hz,2H),0.86(d,J=4.2 Hz,2H);HR-MS(ESI-TOF)m/z:Calcd for C29H27N5O3S{[M+H]+}526.1895,found 526.1907。
N-(5-(2-氨基-1-(苯并[d] [1,3]二恶唑-5-基)-1H-苯并[d]咪唑-6-基)-2-甲氧基吡啶-3-基)环丙烷磺酰胺(G-6):灰白色固体1.80 g,收率65.0%;1H NMR(DMSO-d6,400 MHz)δ:8.16(s,1H),7.74(s,1H),7.28(q,J=8.4 Hz,2H),7.12(d,J=8.5 Hz,2H),6.99~6.95(m,2H),6.16(s,2H),3.92(s,3H),0.98~0.81(m,5H);HR-MS(ESI-TOF)m/z:Calcd for C23H21N5O5S{[M+H]+}480.1321,found 480.1336。
N-(5-(2-氨基-1-(1-甲基-1H-吡唑-3-基)-1H-苯并[d]咪唑-6-基)-2-甲氧基吡啶-3-基)环丙烷磺酰胺(G-7):白色固体1.65 g,收率64.5%;1H NMR(DMSO-d6,400 MHz)δ:8.25(s,1H),7.94(s,1H),7.84(s,1H),7.40(s,1H),7.33(s,2H),6.65(s,1H),3.95(s,3H),3.94(s,3H),1.10(t,J=6.8 Hz,1H),0.96(d,J=7.3 Hz,2H),0.91(s,2H);HR-MS(ESI-TOF)m/z:Calcd for C20H21N7O3S{[M+H]+}440.1485,found 440.1499。
N-(5-(2-氨基-1-(哌啶-4-基)-1H-苯并[d]咪唑-6-基)-2-甲氧基吡啶-3-基)环丙烷磺酰胺(G-8):灰白色固体1.78 g,收率66.9%;1H NMR(DMSO-d6,400 MHz)δ:8.25(s,1H),7.82(d,J=2.1 Hz,1H),7.48(s,1H),7.24(d,J=8.2 Hz,1H),7.18(d,J=8.4 Hz,1H),4.37(t,J=12.2 Hz,1H),3.94(s,3H),3.55(d,J=4.3 Hz,2H),3.23(t,J=12.5 Hz,2H),2.72~2.67(m,1H),1.80(d,J=11.2 Hz,2H),1.22(s,1H),0.92(dd,J=21.3 Hz,5.7 Hz,5H);HR-MS(ESI-TOF)m/z:Calcd for C21H26N6O3S{[M+H]+}441.2703,found 441.2966。
N-(5-(2-氨基-1-(4-吗啉代苯基)-1H-苯并[d]咪唑-6-基)-2-甲氧基吡啶-3-基)环丙烷磺酰胺(G-9):白色固体1.78 g,收率:58.9%;1H NMR(DMSO-d6,400 MHz)δ:8.09(s,1H),7.70(s,1H),7.35(d,J=8.4 Hz,2H),7.31~7.24(m,2H),7.15(d,J=8.6 Hz,2H),6.91(s,1H),3.90(s,3H),3.77(s,4H),3.21(s,4H),1.22(s,1H),0.87(d,J=21.7 Hz,4H);HR-MS(ESI-TOF)m/z:Calcd for C26H28N6O4S{[M+H]+}521.1949,found 521.1966。
2 结果与讨论
2.1 3的合成
考察不同还原体系对3合成的影响,结果见表1。由表1可知,选择Fe-NH4Cl体系时,反应时间最短,收率最高,达到93.4%。
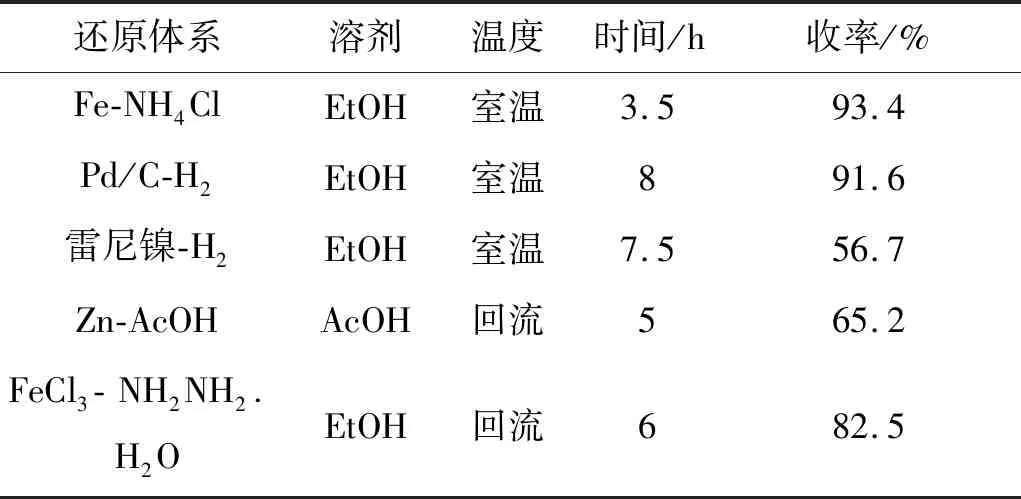
表1 还原体系对3收率的影响Table 1 Effect of reduction systeHR-MS on the yield of compound 3
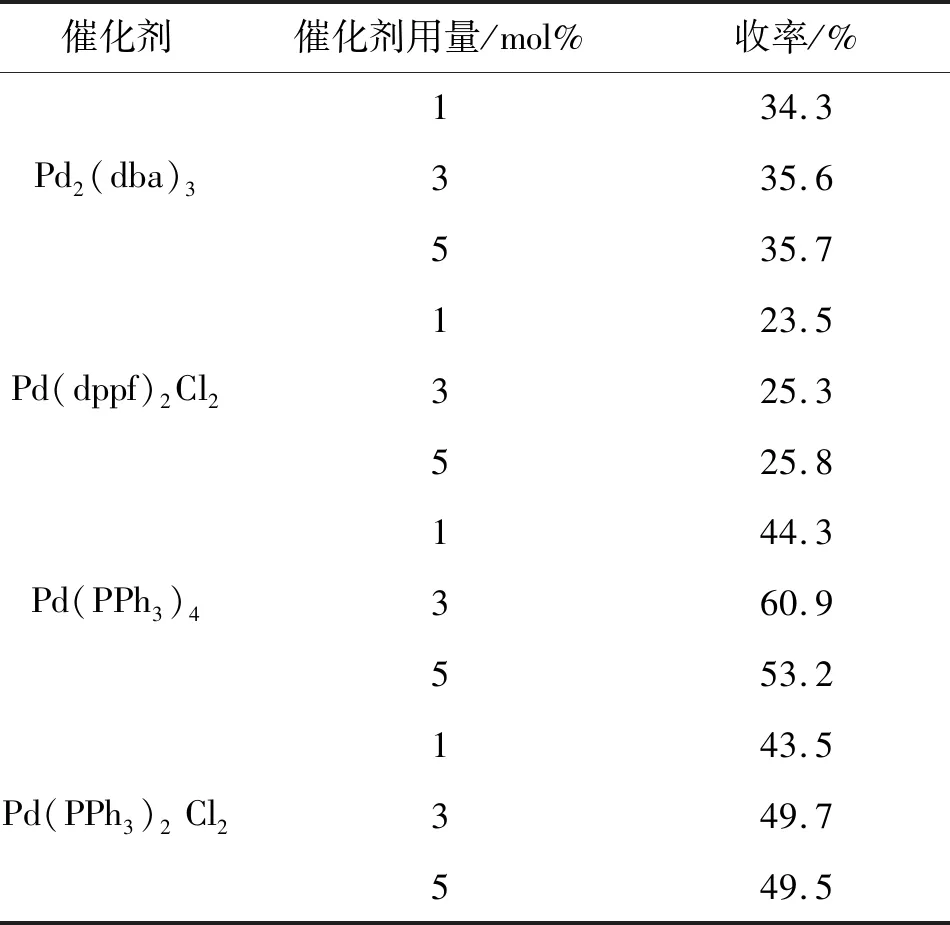
表 3 催化剂对12收率的影响Table 3 Effect of catalyst on yield of compound 12
2.2 4的合成
4的合成是本反应较难的一步,由于直接往反应溶剂里通SO2气体有一定危险性,故实验选择将SOCl2滴加到0 ℃水中,保持混合物温度为0 ℃,反应17 h作为磺酰基的来源。将SOCl2滴加到水中反应17 h,这个过程温度对反应收率影响很大,对温度有很严格的要求。因为不同的温度SO2在水中溶解度有差异,故实验考察了温度对4收率的影响,结果见表2。由表2可知,将SOCl2滴加到0 ℃水中时,收率最高达80.6%,因此选择将SOCl2滴加到0 ℃的冰水中,反应17 h作为磺酰氯化的反应条件。

表2 温度对4收率的影响Table 2 Effect of temperature on the yield of compound 4
2.3 12的合成
在合成12时,发现选用不同的催化剂体系以及用量对反应会产生影响,结果见表3。由表3可知,以Pd(PPh3)4为催化剂,用量为3 mol%时,收率最高为60.9%。
以5-溴-2-甲氧基吡啶、吗啉、2-氟苯胺和联硼酸频那醇酯等为原料,经过硝化、磺酰化、硼酸酯化和Suzuki-Miyaura等反应得到目标GSK-A1,对反应条件进行了优化,优化后各步反应操作条件简单温和、产率较高、后处理简便,总收率达19.33%,便于后续的中试放大研究。在GSK-A1合成工艺基础上,设计并合成了9个新的衍生物,且得到无合成文献报道的中间体5-溴-2-甲氧基-3-磺酰氯吡啶,为GSK-A1衍生物的深入研究奠定了基础。
猜你喜欢
化学与生物工程(2022年7期)2022-08-03
长江大学学报(自科版)(2021年3期)2021-06-01
世界农药(2020年12期)2021-01-04
中华养生保健(2020年3期)2020-11-16
武汉工程大学学报(2020年1期)2020-04-28
农药科学与管理(2019年8期)2019-11-23
农药科学与管理(2019年8期)2019-11-23
今日农业(2019年11期)2019-08-13
农药科学与管理(2019年12期)2019-05-20
成长·读写月刊(2017年3期)2017-04-08
