
高稳定性纳米Y-TiOPc的制备及其应用性能研究
2021-04-01 07:46:30韩明明张亚琴董晓菲刘红丽王世荣李祥高
中国材料进展 2021年3期
刘 流,韩明明,张亚琴,董晓菲,刘红丽,王世荣,李祥高
(天津大学化工学院 天津市功能精细化学品技术工程中心,天津 300350)
1 前 言
Y型酞菁氧钛(Y-TiOPc)具有无毒、光热稳定性好、对600~900 nm波长的光吸收性强和载流子产生效率高等优点。作为红光和近红外光响应的材料,Y-TiOPc在激光有机光导(OPC)[1-3]、有机光电探测(OPD)[4, 5]、有机薄膜晶体管(OTFT)[6]、有机太阳能电池(OSC)[7]和有机发光二极管(OLED)[8]等领域具有重要的应用前景,是有机光电子材料领域的研究热点之一。
TiOPc晶体属于分子晶体[9],由于Ti==O基团垂直于酞菁环(Pc)环平面,具有较强的偶极矩[10, 11],使TiOPc形成不同于完全平面分子的堆积方式。同时,TiOPc晶体中相邻分子间的弱π—π相互作用导致同质多晶现象,使其表现出可变化的光物理和光化学性质。TiOPc晶体主要有α,β和Y 3种晶型,α-晶体为三斜晶系,β-晶体和Y-晶体均为单斜晶系[12]。Y-晶型的晶胞中TiOPc分子呈对角线堆积方式(图1),这种方式最有利于电荷从Pc环向Ti转移,使Y-晶型光量子产率可达90%以上[13]。相较于低光量子产率的α-晶型和β-晶型,Y-晶型稳定性较差,为了得到高光量子产率材料,研究稳定Y-TiOPc的制备方法成为关键问题。
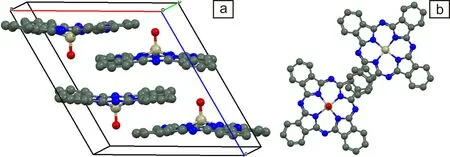
图1 Y-TiOPc分子在晶胞中的堆积方式:(a)主视图,(b)俯视图Fig.1 Schematic diagrams of packing form of Y-TiOPc in a crystal cell: (a) front view, (b) top view
王远等[14]利用胶体-溶液介导相转变法,以浓硫酸溶解TiOPc,将得到的溶液滴加到低温甲醇溶液中析出沉淀,再用1,2-二氯乙烷(1,2-C2H4Cl2)将纳米TiOPc萃取出来,制备了平均粒径为3.4 nm的Y-TiOPc,以其制备的单层OPC器件显示出了较高的光敏性(E1/2=0.34 μJ/cm2);李小龙等[1, 4]以邻二氯苯(o-C6H4Cl2)/水形成的微乳液-相转移体系制备了平均粒径为2.3 nm的Y-TiOPc,以其制备的双层OPC器件的E1/2提高到了0.064 μJ/cm2,制备的OPD器件在电场强度为15 V/μm、入射光功率为0.1 μW/cm2(780 nm)的条件下,外量子产率EQE=354200%,光响应度R=2227 A/W,比探测率D*=3.1×1014;乐园等[15]用超重力内/外循环旋转填充床联用的方法制备了平均粒径为6.0 nm的Y-TiOPc,且以其制备的双层光导器件的E1/2=0.166 μJ/cm2。工业化制备Y-TiOPc最常用的方法为浓硫酸酸糊法,即将TiOPc/H2SO4溶液滴加至冰水混合物中,使TiOPc结晶析出,洗涤、干燥后得到无定型TiOPc粉末,再将粉末加入o-C6H4Cl2/H2O混合物中调节晶型,得到Y-TiOPc。李小龙等[1]将此方法制备的Y-TiOPc作为载流子产生材料制备多层有机光电导器件,器件性能为:充电电位V0=-706.80 V,暗衰速率Rd=-25.60 V/s,残余电位Vr=-39.10 V,光敏性E1/2=0.116 μJ/cm2。李云梦等[16]将此方法制备的Y-TiOPc作为载流子产生材料制备单层OPC器件,器件性能为:V0=550.90 V,Rd=-36.82 V/s,Vr=134.73 V,E1/2=0.290 μJ/cm2。以上方法都是以浓H2SO4作为溶解TiOPc的溶剂,工艺过程缺乏安全性和环境友好性,更为严重的是制备的Y-TiOPc在溶剂诱导或机械力作用下易转变为β-TiOPc,导致器件性能下降。孙煜伟等[17]将TiOPc/三氟乙酸(TFA)/1,2-C2H4Cl2混合溶液滴加至0 ℃的去离子水中析出TiOPc,过滤、洗涤后,将湿滤饼加入至50 ℃的o-C3H4Cl2中调节晶型,得到Y-TiOPc,以其制备的多层光导器件性能为:V0=-828.70 V,Rd=-20.30 V/s,Vr=-33.70 V,E1/2=0.090 μJ/cm2。但是,此方法制备的产物需要用大量有机溶剂在极稀的浓度下分离,不利于实用化制备。
本文以TFA为溶解介质,将TiOPc/TFA溶液滴加到不同体积分数的乙醇溶液中析出沉淀,将得到的沉淀加入在强剪切作用下形成的1,2-C2H4Cl2/H2O微乳液中,在不同温度条件下调节晶型,从而得到晶粒尺寸小、结晶度高且晶型稳定的纳米Y-TiOPc,并用其制备出高性能的近红外光电导器件。
2 实 验
2.1 实验材料
TiOPc粗品和N,N,N′,N′-四(4-甲基苯基)-1,1′-联苯-4,4′-二胺(S100)为实验室合成;丁酮(MEK)和环己酮(CYC)购自天津市江天化工技术有限公司;甲醇、乙醇、正丁醇、环己烷、二氯甲烷和1,2-二氯乙烷购自利安隆博华(天津)医药化学有限公司;邻二氯苯购自日本kureha株式会社;TFA购自天津科密欧化学试剂有限公司。聚乙烯醇缩丁醛(PVB,分子量为90 000~120 000)和聚酰胺(PA,分子量为15 000~30 000)、聚碳酸酯(PC,分子量为45 000)分别购自美国阿拉丁公司和西格玛奥德里奇公司。所有溶剂蒸馏后使用。
2.2 纳米Y-TiOPc及光电导器件的制备
2.2.1 1,2-C2H4Cl2/H2O微乳液的制备
制备1,2-C2H4Cl2/H2O微乳液最简单的方法就是通过机械搅拌为1,2-C2H4Cl2和H2O提供足够的剪切力以达到乳化效果。测试120 mL体积比为2∶1的1,2-C2H4Cl2/H2O体系的电导率与剪切速率的关系,如表1所示。当剪切速率由100增至200 r/min时,该体系的电导率迅速由263降低至14 μS/cm,说明1,2-C2H4Cl2/H2O形成的微乳液类型为油包水(W/O)型;当剪切速率为900 r/min时,1,2-C2H4Cl2/H2O体系的电导率降至1 μS/cm(1,2-C2H4Cl2的电导率为3×10-4μS/cm),再增大剪切速率时体系的电导率也不再变化,故此剪切速率下已经形成了稳定的W/O型微乳液。因此,本实验的微乳液是以体积比为2∶1的1,2-C2H4Cl2和H2O在剪切速率为900 r/min的条件下制备的。

表1 1,2-C2H4Cl2/H2O体系的电导率与剪切速率的关系
2.2.2 纳米Y-TiOPc的制备
在0 ℃下,将5.0 g TiOPc粗品完全溶解于100 mL TFA中,将得到的溶液分别滴加到800 mL体积分数(下同)为0、25%、50%、75%和100%的乙醇溶液(0 ℃)中,在900 r/min的搅拌速率下使TiOPc结晶析出;经过滤、洗涤后,将得到的滤饼直接转移至1,2-C2H4Cl2/H2O微乳液中,分别在10,20,30,40和50 ℃下调节晶型2 h;然后,滴加乙醇破乳,使TiOPc析出;静置分层后,下层1,2-C2H4Cl2相呈蓝色,上层H2O相为无色透明;分液后用去离子水洗涤有机相,直至洗涤液电导率低于2 μS/cm为止;将洗涤液过滤,得到的滤饼用环己烷分散后真空冷冻干燥,最后得到Y-TiOPc蓝色粉末。另外,浓硫酸酸糊法制备Y-TiOPc的步骤详见文献[1]。
2.2.3 Y-TiOPc纳米粒子分散液的配制
在球磨瓶中,将0.1 g PVB完全溶解在10 mL等体积比的MEK/CYC混合溶剂中,然后加入0.1 g制备的纳米Y-TiOPc,超声波作用30 min后加入50 g直径为1 mm的ZrO2珠,在对辊机上球磨分散2 h(转速为360 r/min),经聚丙烯膜(孔径为10 μm)过滤,得到蓝色的Y-TiOPc/MEK/CYC/PVB分散液。
2.2.4 OPC器件的制备
Y-TiOPc的光电导性能需要通过OPC器件进行表征,器件结构由铝箔基底、预涂层(UCL)、载流子产生层(CGL)和空穴传输层(HTL)构成,如图2所示。
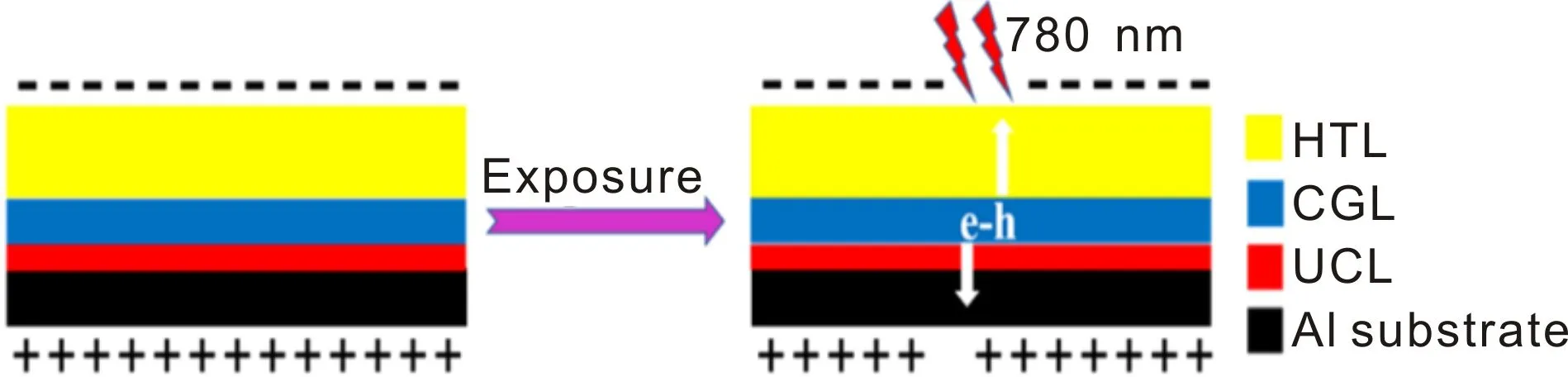
图2 激光有机光导(OPC)器件结构示意图及光致诱导放电过程Fig.2 Structural schematic diagram and photoinduced discharging processe of OPC device
将2.50 g PA完全溶解在50 mL体积比为4∶1的甲醇/正丁醇混合溶液中,然后涂布在洁净、干燥的铝箔基底上,在80 ℃下烘干后得到UCL;将制备的Y-TiOPc/MEK/CYC/PVB分散液涂布在UCL上,背光晾干后在90 ℃下干燥30 min后得到CGL;将3.60 g PC完全溶解于25.5 mL的1,2-C2H4Cl2中,然后再向该溶液中加入2.52 g S100并使其完全溶解,涂布于CGL上,室温避光干燥后再在100 ℃下干燥2 h得到HTL,由此得到的OPC器件用于测试分析Y-TiOPc的光电导性能。
2.3 测试与表征
采用Mini Flex 600型X射线衍射仪(XRD)(日本Rigaku公司)对TiOPc进行物相分析。测试时采用Cu靶,扫描电压为40 KV,扫描电流为15 mA,扫描范围为5°~35°,扫描速度为7°/min,步长为0.02°,连续扫描。采用Q50型热重分析仪(美国TA仪器公司)对两种方法制备的纳米Y-TiOPc进行分析,测试条件为:10 mg样品,N2氛围,25~300 ℃,升温速率为10 ℃/min。本实验的接触角(CA)测试采用JC2000D型静态接触角测量仪(上海中晨数字技术设备有限公司)。采用FTIR-650型红外光谱仪(天津港东科技发展股份有限公司)对两种方法制备的纳米Y-TiOPc的纯度进行鉴定,具体是将3 mg 左右的待测样品与0.2 g KBr充分研磨混合后,压片,测试其红外吸收光谱。
利用UV2600型紫外可见分光光度计(上海天美科学仪器有限公司)对Y-TiOPc分散液的紫外-可见吸收光谱进行测量。分散液中Y-TiOPc纳米粒子的粒径及其分布使用DelsaTMNano C型纳米粒度仪(美国贝克曼库尔特公司)进行测量。采用Turbiscan LABTM型分散稳定性测试仪(法国Formulaction公司)对Y-TiOPc分散液稳定性进行表征,此方法是测试分散液的透射率随静置时间的变化值,透射率随时间的变化越大,分散液稳定性越差。采用Dimension Icon型原子力显微镜(德国布鲁克公司)对Y-TiOPc分散液的表面形态进行表征:将2~3滴Y-TiOPc/MEK/CYC/PVB分散液滴加在以2000 r/min旋转的玻璃片(2×2 cm2)上,烘干后进行测试。
OPC器件的光电导性能:采用数字化改造的SP-428型静电纸分析仪(日本Kawaguchi电气株式会社)测试其光致放电曲线(photoinduced discharging curve,PIDC),得到OPC器件的光电导性能参数。所采用的单色光波长为780 nm。
3 结果与讨论
通常,TiOPc的提纯方法是将粗品溶解在浓硫酸中,然后在冰水混合体系中析出TiOPc,达到提纯的效果,通过控制搅拌速度等析出条件获得TiOPc纳米粒子。然而,溶解1 g TiOPc需要约40 mL浓硫酸[1],用量很大,而且当浓硫酸溶液滴加到水中会瞬时产生局部过热,导致TiOPc分解生成邻苯二甲酰亚胺和磺化产物等杂质,制备条件严苛。TFA是有机强酸,沸点较低,对TiOPc有良好的溶解性,而且与水或醇混溶时放热小,且可以回收再利用[17]。因此,本实验选择TFA作为溶剂制备纳米Y-TiOPc。
3.1 乙醇溶液浓度对TiOPc晶型的影响
将TiOPc/TFA溶液分别滴加在不同浓度的乙醇溶液中,析出沉淀并经过晶型调节(35 ℃)后得到TiOPc纳米粉末。采用XRD对其物相进行分析,并计算其结晶度(半峰宽>1°视为非晶峰),如图3a和3b所示。当乙醇含量为0时,得到的TiOPc的主要特征衍射峰为7.4°、10.3°、16.4°和24.2°,即为α-TiOPc,其分子在晶胞中的排列方式如图3c所示;当乙醇含量为25%时,产物在9.6°、24.1°和27.3°呈现出Y-TiOPc的特征衍射峰;当乙醇含量增加到50%时,Y-TiOPc的结晶度达到91.68%,但当乙醇含量继续增加,甚至达到100%时,虽然得到的产物仍以Y-TiOPc为主,但结晶度有所下降,并出现α-TiOPc的衍射峰。这表明,沉淀剂的组成对TiOPc分子的堆积方式有很大的影响,在水中沉淀析出后经过微乳液体系的调节只能得到α-晶型,而在乙醇与水的混合体系沉淀析出后经微乳液体系调节可以得到结晶度不同的Y-TiOPc,但在纯乙醇中沉淀时,多晶体中产生的α-晶型经过微乳液体系的调节也不能完全转变为Y-晶型。
3.2 调节温度对TiOPc晶型的影响
TiOPc在体积分数为50%的乙醇溶液中析出后分别经不同温度的1,2-C2H4Cl2/H2O微乳液调节晶型,得到的产物的XRD图谱如图4所示,对应的特征衍射峰的峰强比见表2。可见,调节温度对TiOPc晶型也有较大的影响。当调节温度为10和20 ℃时,α-TiOPc在特征衍射峰7.6°处的峰强比分别高达37.4%和23.9%,说明在此温度下,α-TiOPc并不能完全转化为Y-TiOPc;随着调节温度的升高,7.6°处的峰强基本呈降低趋势。当调节温度为30 ℃时,Y-TiOPc的特征衍射峰9.6°处的峰强比为33.3%,且该温度下的结晶度为93.82%;当调节温度为40 ℃时,9.6°处的峰强比增至44.9%,但制备的Y-TiOPc光电活性较差[17];当调节温度升至50 ℃时,β-TiOPc的特征衍射峰10.4°和26.2°出现。随着晶型调节体系能量的增大,晶体可以发生位移式转变,形成热力学更稳定的晶型。综上,本实验选择的晶型调节温度为30 ℃。

图3 不同浓度的乙醇溶液中析出得到的酞菁氧钛(TiOPc)的XRD图谱(a)和结晶度(b);α-TiOPc分子在晶胞中的排列方式示意图(c)Fig.3 XRD patterns (a) and crystallinity (b) of TiOPc precipitated in solutions with different volume fractions; the schematic diagram of α-TiOPc molecules arranged in a unit cell (c)
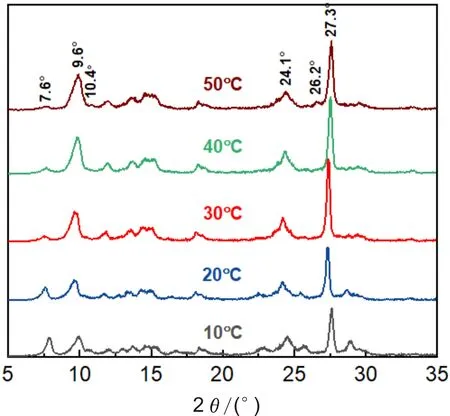
图4 不同晶型调节温度下制备的TiOPc的XRD图谱Fig.4 XRD patterns of TiOPc prepared at different adjustment temperatures
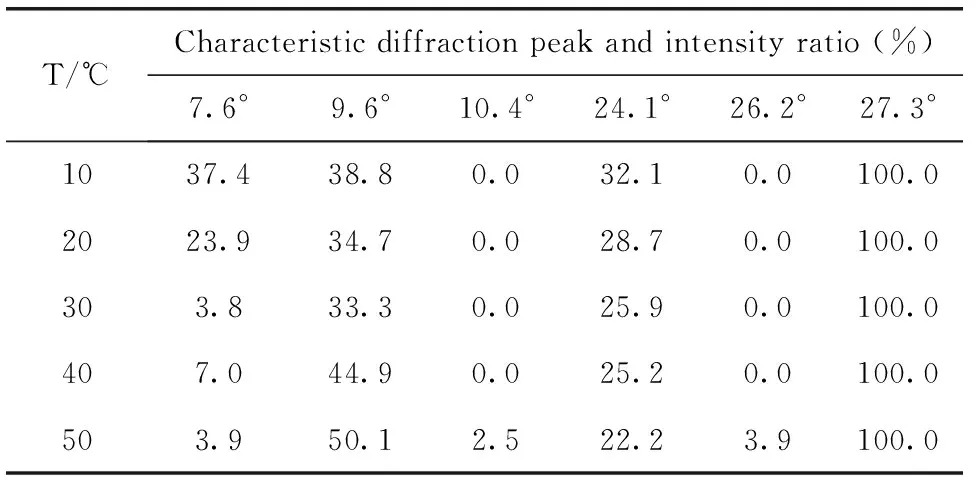
表2 不同调节温度下制备的TiOPc的特征衍射峰及峰强比
TFA酸糊法和经典浓硫酸酸糊法处理后,经晶型调节制备的Y-TiOPc的XRD图谱如图5a所示。对比发现,I9.6°/I27.3°衍射峰强度比,前者为33.3%、后者为47.7%,表明TFA酸糊法制备Y-TiOPc,经1,2-C2H4Cl2/H2O微乳液体系进行晶型调节更优先生长(222)晶面,光电活性更高[17]。两种方法制备的Y-TiOPc的粒径分布如图5b和5c所示,TFA酸糊法制备的Y-TiOPc平均粒径为2.86 nm,而浓硫酸酸糊法制备的Y-TiOPc平均粒径为30.19 nm。本实验采用的TFA酸糊法可获得更小粒径的Y-TiOPc。

图5 TFA体系和浓硫酸体系制备的Y-TiOPc的XRD图谱(a);三氟乙酸(TFA)体系(b)和浓硫酸体系(c)制备的Y-TiOPc的粒径分布情况Fig.5 XRD patterns of Y-TiOPc prepared by TFA system and H2SO4 system (a); particle size distribution of Y-TiOPc prepared by TFA system (b) and H2SO4 system (c)
两种方法制备的Y-TiOPc的热重(TG)和差示扫描量热(DSC)曲线见图6。分析可知,TFA体系制备的产物只在100~200 ℃ 附近有明显的失重,这是Y-TiOPc表面H2O分子以及晶胞内Ti4+上结合的H2O分子蒸发所致[18, 19]。而浓硫酸体系产物在这个温度范围没有失重,说明分子中没有结晶水形成。但是,它在250℃附近有明显的失重和吸热峰,失重率达到1%。浦冰叶等[20]认为这是由于用浓硫酸提纯时,局部过热导致少量的TiOPc分解产生邻苯二甲基亚酰胺。
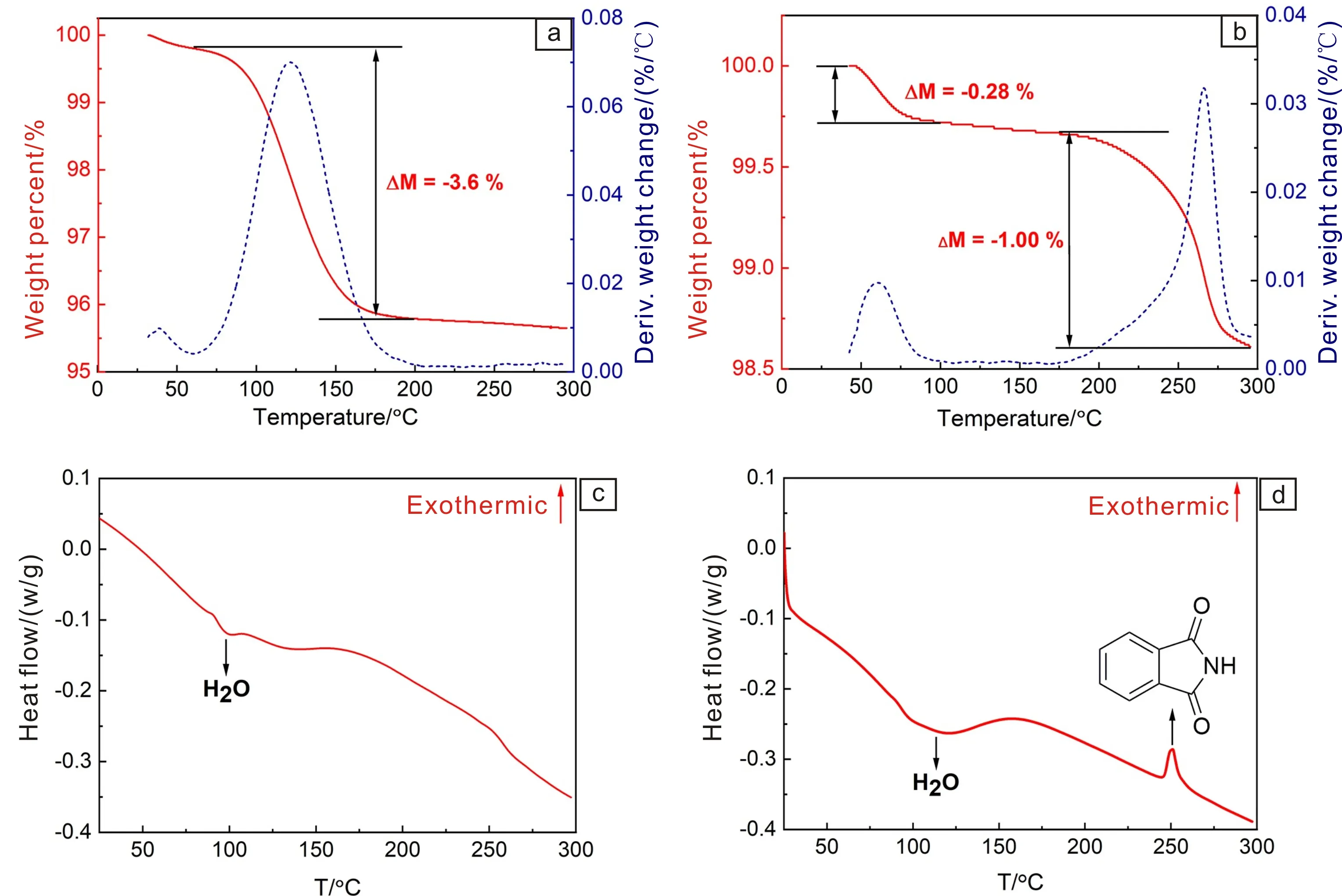
图6 Y-TiOPc样品的TG(a, b)和DSC(c, d)曲线:(a, c)TFA体系, (b, d)浓硫酸体系Fig.6 TG (a, b) and DSC (c, d) curves of Y-TiOPc: (a, c) TFA system, (b, d) H2SO4 system
TFA体系和浓硫酸体系制备的Y-TiOPc和H2Pc的红外光谱(IR)如图7所示,浓硫酸体系制备Y-TiOPc和H2Pc在1007 cm-1处有中等强度的吸收峰,这是由于H2Pc中N—H键伸缩振动所致,而TFA体系制备的Y-TiOPc无此峰产生,但在964 cm-1处出现的吸收峰对应的是Ti==O键摇摆伸缩振动[21]。可见,本实验的提纯和晶型转化方法可获得纯度更高的Y-TiOPc。
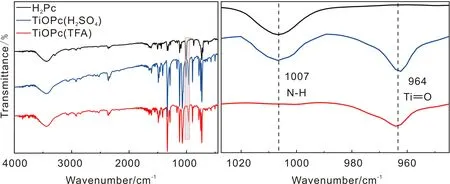
图7 TFA体系和浓硫酸体系制备的Y-TiOPc和H2Pc的红外光谱(IR)图Fig.7 IR spectra of Y-TiOPc and H2Pc prepared by TFA system and H2SO4 system
3.3 Y-TiOPc的晶型稳定性
Y-TiOPc具有优异的光电转化性能,但其稳定性较差,在机械力作用或溶剂诱导下易转化为稳定性更高的β-TiOPc,故本文研究了Y-TiOPc的晶型稳定性。
TFA和浓硫酸体系制备的Y-TiOPc在MEK/CYC/PVB混合溶剂中分别经超声波振动和球磨分散作用不同时间后,得到的产物的XRD图谱,如图8所示。浓硫酸体系制备的Y-TiOPc在超声波作用1 h后,在10.4°和26.2°处出现β-晶型的特征衍射峰,峰强比分别为3.1%和8.7%,而且随着超声波作用时间的延长,β-晶型的特征峰越来越明显,作用4 h后,这两处的β-晶型的衍射峰峰强比分别增加到8.6%和32.9%。而TFA体系制备的Y-TiOPc在超声波作用下表现出非常稳定的特性,4 h后仍保持完全的Y-晶型。在球磨作用下,浓硫酸体系制备的Y-TiOPc在1 h时也出现β-晶型的特征衍射峰,峰强比分别为13.3%和54.4%;作用2 h后,峰强比分别增加到50.5%和100%,作用4 h后则完全转化为β-晶型,Y-晶型的衍射峰消失。而TFA体系制备的Y-TiOPc在球磨作用下2 h内仍为Y-晶型,只有达到4 h后,才在26.2°处出现了很小的β-晶型衍射峰,峰强比仅为4.1%。因此,本实验的提纯和晶型转化方法可获得晶型更稳定的Y-TiOPc。

图8 超声波(a, b)和球磨分散(c, d)分别作用不同时间后,Y-TiOPc的XRD图谱:(a, c)TFA体系,(b, d)浓硫酸体系Fig.8 XRD patterns of Y-TiOPc respectively treated by ultrasonication (a, b)and ball milling(c, d) with different time: (a, c) TFA system, (b, d)H2SO4 system
两种方法制备的纳米Y-TiOPc在MEK/CYC/PVB分散液中的紫外-可见光吸收光谱,如图9所示。它们在340~400(B带)、692(TiOPc单分子吸收峰)和720~850 nm(TiOPc聚集态吸收峰,Q带)均具有强吸收峰[22, 23]。结果表明,两种方法制备的Y-TiOPc纳米粒子在MEK/CYC/PVB分散液中具有一定的溶解性,且存在单分子状态。但TFA体系制备的Y-TiOPc在分散液中的最大吸收峰(780 nm)较浓硫酸体系的(830 nm)发生了明显的蓝移,表明TFA体系制备的Y-TiOPc粒子的分子之间的π—π作用更强、堆积更紧密,导致处于该能态的价带电子需要更大的能量才能被激发,这也是其晶型更稳定的原因之一。另外,图3a也可以证实通过与H2O分子的氢键作用,TFA体系制备的产物含有类似“结晶水”的结构,使得π—π作用增强,分子堆积更紧密,增强了Y-TiOPc的稳定性。
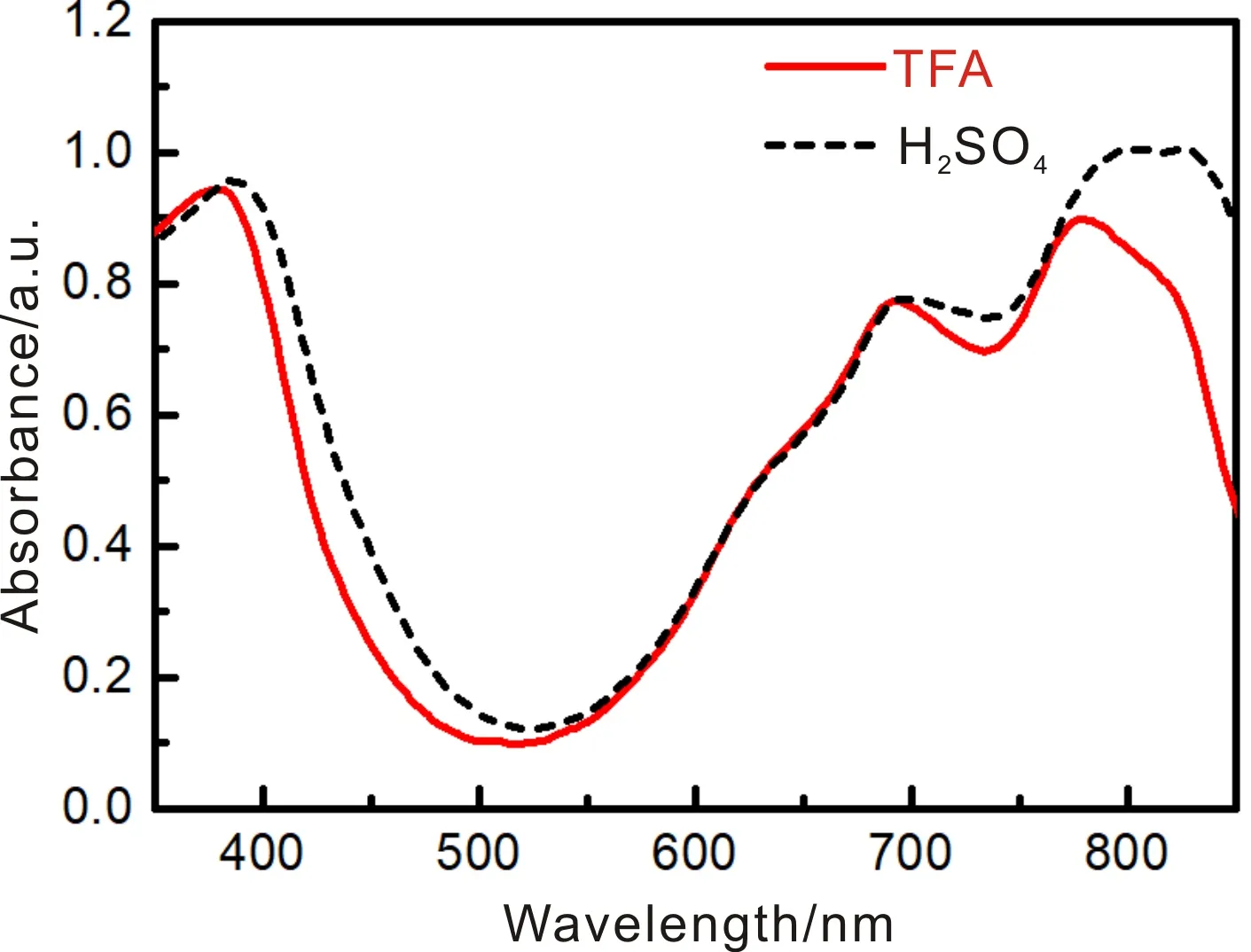
图9 TFA体系和浓硫酸体系制备的Y-TiOPc在分散液中的紫外-可见光吸收光谱Fig.9 UV-visible absorption spectra of Y-TiOPc dispersion prepared by TFA system and H2SO4 system
为进一步探究TiOPc晶型稳定性差异的机理,分别测量了经1,2-C2H4Cl2和o-C6H4Cl2提纯后TiOPc的接触角,并配制了0.04 g/L的TiOPc的1,2-C2H4Cl2和o-C6H4Cl2的胶体,在650 nm波长的红色激光照射下,观察其丁达尔效应(图10)。1,2-C2H4Cl2和o-C6H4Cl2与TiOPc的接触角分别为5.4°和13.3°,而且相同浓度下TiOPc/1,2-C2H4Cl2胶体中的丁达尔效应更明显。这说明TiOPc粗品在1,2-C2H4Cl2中的分散性更好。由于1,2-C2H4Cl2与TiOPc具有良好的相溶性,所以TiOPc分子与1,2-C2H4Cl2分子的相互作用更强,尤其在(222)晶面上,1,2-C2H4Cl2分子吸附导致(222)晶面表面张力减小,促进了TiOPc分子在此晶面上的生长。同时,本实验采用的W/O型微乳液,在晶型调节过程中,大量的TiOPc分子存在于水相和1,2-C2H4Cl2相界面处,更易与H2O分子生成分子间氢键,使得π—π作用增强,分子堆积更为紧密,晶型稳定性增强。而浓硫酸酸糊法制备Y-TiOPc时需要将干燥后的无定型TiOPc粉末置于o-C6H4Cl2/H2O微乳液中,TiOPc的二次团聚及其与o-C6H4Cl2的相溶性较差,必然导致部分TiOPc分子不能与H2O相、o-C6H4Cl2相完全作用,从而不利于其优先生长(222)晶面,也不利于其与H2O分子形成分子间氢键,导致分子堆积不紧密,晶型稳定性差。

图10 不同溶剂与TiOPc粗品的接触角及丁达尔效应:(a)1,2-C2H4Cl2,(b)o-C6H4Cl2Fig.10 Contact angle and Tindal Effect between different solvents and crude TiOPc: (a) 1,2-C2H4Cl2, (b) o-C6H4Cl2
3.4 Y-TiOPc的分散稳定性
分别将两种方法制备的Y-TiOPc纳米粒子配制成分散液,静置30 d观察分散液顶部透射率的变化。TFA体系制备的Y-TiOPc的分散液透射率的变化率仅为0.53%,而浓硫酸体系制备的Y-TiOPc的分散液透射率的变化率则达到了1.89%,可见后者粒子更容易聚集和沉降,分散稳定性较差。
分散液中PVB链上的—OH可以与Y-TiOPc的Ti==O形成氢键,从而吸附在Y-TiOPc纳米粒子表面增大分散液中固体颗粒的粒径,利用动态光散射原理分别测试两种方法制备的Y-TiOPc在MEK/CYC/PVB分散液中的粒径分布,如图11a所示,TFA体系与浓硫酸体系制备的Y-TiOPc的平均粒径分别为73.2和96.7 nm。由于浓硫酸体系制备的Y-TiOPc在分散液中的粒径更大,在重力作用下,粒子的沉降速率相对更快,分散液稳定性较差。另外,MEK/CYC/PVB分散液与TFA体系、浓硫酸体系制备的Y-TiOPc表面的接触角分别为7.9°和15.6°(图11b和11c)。可见TFA体系制备的Y-TiOPc对分散液具有更好的相容性,这也使其更易在分散液中分散。
将两种体系制备的Y-TiOPc/MEK/CYC/PVB分散液涂敷在载玻片上进行观察,得到的AFM三维微观形貌图如图12所示。在扫描范围为2.5×2.5 μm2、观察尺度为75.0和140.0 nm的条件下,薄膜的均方根粗糙度Rq分别为25.3和33.6 nm。可见TFA体系制备的Y-TiOPc具有更好的薄膜均匀性,这也是分散稳定性提高的结果。
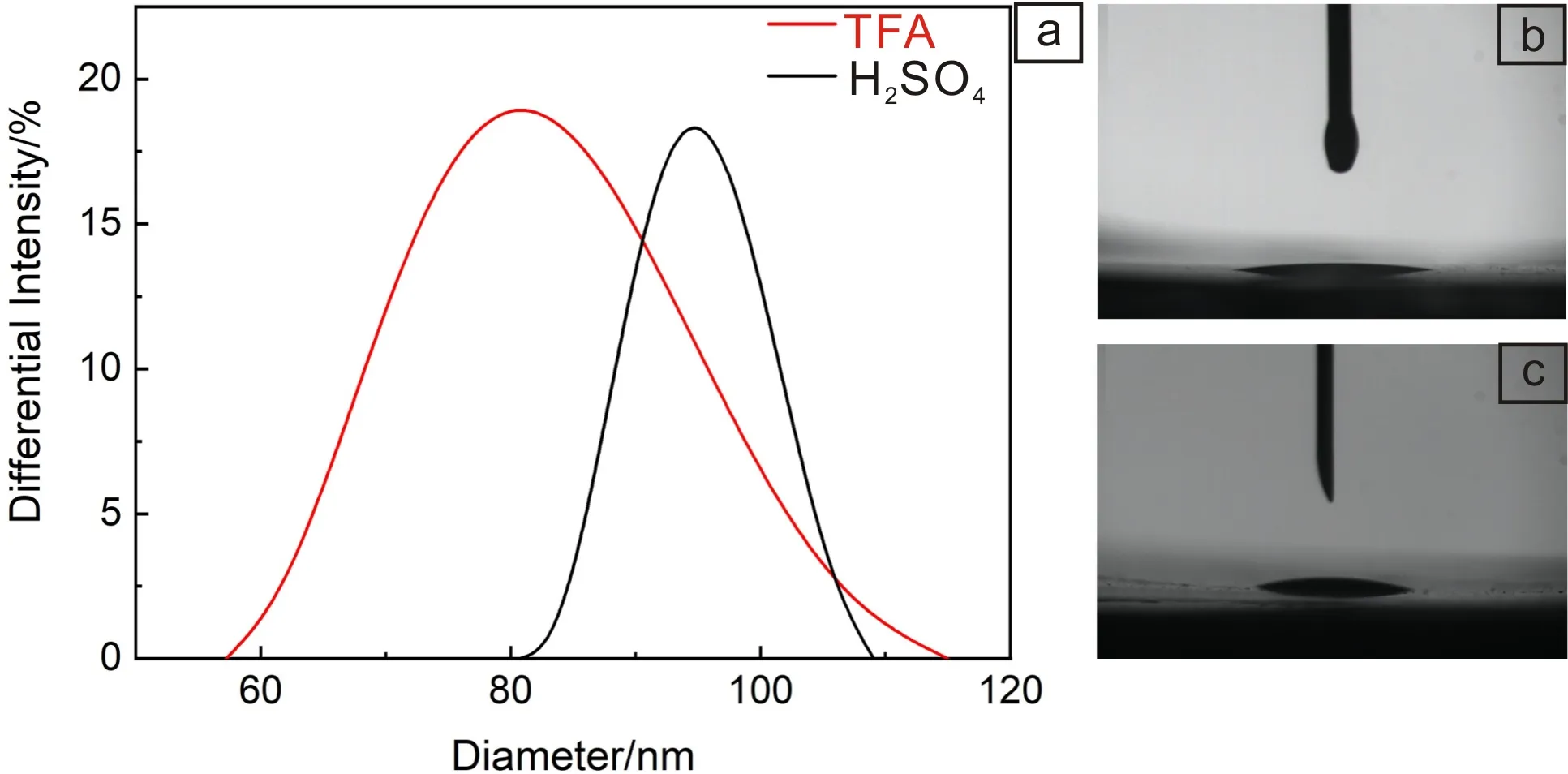
图11 Y-TiOPc在分散液中的粒径分布(a);分散液分别与由TFA体系(b)和浓硫酸体系(c)制备Y-TiOPc的表面接触角Fig.11 Particle size distribution of Y-TiOPc in dispersion (a); surface contact angle between dispersion and Y-TiOPc prepared by TFA system (b) and H2SO4 system (c)
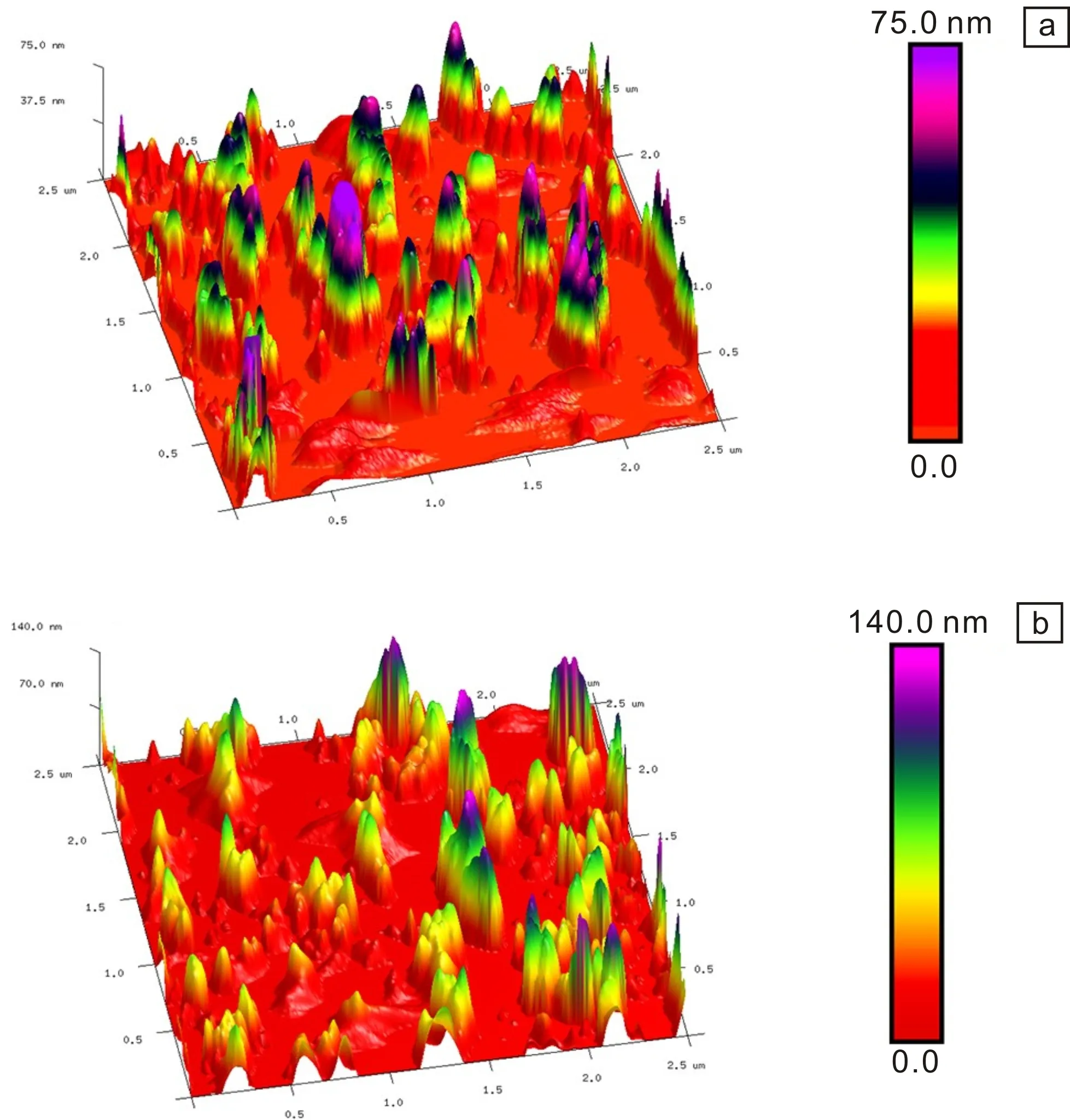
图12 Y-TiOPc@PVB薄膜的AFM三维微观形貌图:(a)TFA体系,(b)浓硫酸体系Fig.12 3D AFM micrographs of Y-TiOPc@PVB films: (a) TFA system,(b) H2SO4 system
3.5 Y-TiOPc的光电导性能
以制备的纳米Y-TiOPc为载流子产生材料、S100为空穴传输材料、醇溶性PA为阻挡材料制备的OPC器件表征Y-TiOPc的光电导性能,在780 nm激光束扫描下测试其光电导特性,得到的PIDC曲线如图13所示。V0代表暗态下电晕放电后光电导器件表面的电位,数值越大电荷接受能力越强;Rd是器件在暗态下表面电位的衰减速率,数值越小电荷保持能力越强;Vr定义为激光束扫描1 s以后器件表面的电位,数值越小器件的光电导能力越强;E1/2定义为器件表面电位降低到1/2时所需要的曝光能量,数值越小光敏性越好。从图中可见,浓硫酸酸糊法制备的Y-TiOPc的器件性能参数为:V0=-729.90 V,Rd=-12.21 V/s,Vr=-41.50 V,E1/2=0.21 μJ/cm2;而TFA体系制备的Y-TiOPc的器件的性能参数为:V0=-878.89 V,Rd=-12.10 V/s,Vr=-14.65 V,E1/2=0.17 μJ/cm2,性能更优。

图13 Y-TiOPc纳米粒子作为载流子产生材料制备的OPC器件的暗衰曲线(a)和光致诱导放电曲线(PIDC)(b)Fig.13 Dark decay curves (a) and PIDCs (b) of OPC devices prepared using Y-TiOPc as carriers generating material
4 结 论
本文采用有机强酸TFA溶解TiOPc粗品,通过在体积分数为50%的乙醇溶液中析出提纯,然后在强剪切力作用下形成的 1,2-C2H4Cl2/H2O微乳液(30 ℃)中调节晶型制备出了晶型稳定性高的纳米Y-TiOPc,其平均粒径为2.86 nm,结晶度达到了93.82%,在丁酮/环己酮体系中超声波或球磨分散作用4 h均保持晶型稳定。改善晶型稳定性的原因是TFA酸糊法使Y-TiOPc优先生长(222)晶面,且Y-TiOPc分子能够与H2O分子生成分子间氢键,使得π—π作用增强,分子堆积更紧密。TFA体系制备的Y-TiOPc纳米粒子在有机溶剂中具有更好的分散稳定性,静置30 d后透射率的变化率仅为0.53%;而且,以其制备的OPC器件的光电导性能优异。
猜你喜欢
青岛科技大学学报(自然科学版)(2023年6期)2023-11-25 17:17:56
陶瓷学报(2020年2期)2020-10-27 02:16:14
中学化学(2017年2期)2017-04-01 12:49:32
中学生数理化·中考版(2017年1期)2017-03-29 19:41:47
中国果菜(2016年9期)2016-03-01 01:28:39
中国塑料(2015年6期)2015-11-13 03:02:34
中国塑料(2015年8期)2015-10-14 01:10:48
中学化学(2015年5期)2015-07-13 07:45:07
中国洗涤用品工业(2015年11期)2015-02-28 19:03:09
济宁医学院学报(2014年4期)2014-08-16 13:44:19
