
胆管癌靶向治疗研究进展
2021-04-01 01:54彭鹏强晓妍田玉伟王慧敏
药学进展 2021年1期
彭鹏,强晓妍,田玉伟,王慧敏
(南京药捷安康生物科技有限公司,江苏 南京210032)
胆管癌(cholangiocarcinoma,CCA)是起源于胆管上皮细胞的恶性肿瘤,临床少见,异质性极高,侵袭性较强且预后差[1]。根据解剖位置的不同,胆管癌最常见的分类为:肝内胆管癌(intrahepatic CCA,iCCA)、肝门部胆管癌(perihilar CCA,pCCA)和远端胆管癌(distal CCA,dCCA),后两者也被合并称为肝外胆管癌(extrahepatic CCA,eCCA)。CCA与胆囊癌(gallbladder cancer,GBC)合并称为胆道癌(biliary tract cancer,BTC)。CCA全球发病率和死亡率近年均呈上升态势[1]。据统计,2019年,全球约有40万CCA新增发病病例,其中,亚洲近28万例[2]。
由于CCA患者(尤其是iCCA)早期无明显临床症状,晚期才会出现盗汗、黄疸、腹腔积液、肝肿大等特异性临床表现,此时疾病经诊断多为中晚期。其中,可经手术/肝移植的患者仅约35%,且术后易复发,5年生存率大约为5% ~ 15%[3]。对于不可手术或不适合局部治疗(如肝动脉介入治疗或射频/微波消融)的CCA患者,化疗和放疗是主要的治疗方式。其中顺铂联合吉西他滨(CisGem)被认为是目前iCCA全身化疗的一线金标准,中位总生存期(median overall survival,mOS)小于1年,预后不理想[4-6]。放疗如外束放疗、立体定向放疗、质子束治疗等虽对CCA有一定的治疗作用,但由于不良反应较大、临床数据较少等原因,目前仍缺乏充分经临床验证的最佳治疗方案。对于进展过快或浸润转移的CCA患者来说,姑息治疗或减轻癌症症状的护理可能是患者唯一的治疗选择。此外,针对CCA的其他治疗方案包括参加临床试验,但由于CCA的异质性强,仅有部分有相对明确生物标记物的患者可以从临床试验中获益。
随着近年来生物检测技术的成熟,CCA潜在的分子病理正在逐步被揭示。文献研究表明[5],CCA均有极高的异质性。在CCA中,有近40%的患者存在潜在的可靶向的基因突变,具有靶向治疗的可能,而针对CCA的精准分子靶向治疗也在当前治疗方法中日渐崭露头角,这其中包括成纤维细胞生长因子受体2(fibroblast growth factor receptor 2,FGFR2)抑制剂、神经营养因子受体酪氨 酸 激 酶(neurotrophin receptor kinase,NTRK)抑制剂、间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)/c-ros原 癌 基 因1(c-ros oncogene 1,ROS1)抑制剂、异柠檬酸脱氢酶1/2(isocitrate dehydrogenases 1/2,IDH1/2)抑制剂、BRAF抑制剂等。目前已经或正在开展的CCA临床试验中,其中绝大部分为靶向治疗药物,本文对主要靶向治疗进展情况进行综述,旨在为CCA药物研发和临床工作提供相关参考。
1 受体酪氨酸激酶抑制剂
尽管因解剖位置和诱发病因的危险因素不同,CCA基因组特征上有显著差异,但受体酪氨酸激酶(receptor tyrosine kinase,RTK)信号通路的异常激活依旧为CCA中较常见的一种异常。虽然表皮生长因子受体(epidermal growth factor receptor,EGFR)抑制剂以及血管内皮生长因子(vascular endothelial growth factor,VEGF)/血管内皮生长因子受体(vascular endothelial growth factor receptor,VEGFR)抑制剂在临床上已证明对化疗无明显联用增效作用[7],但近年来基因测序技术仍提供了很多RTK相关的CCA治疗新靶点。
1.1 成纤维细胞生长因子受体抑制剂
FGFR是一类跨膜酪氨酸激酶受体,其包括FGFR1 ~ FGFR4和无蛋白酪氨酸激酶域的成纤维细胞生长因子受体样1(fibroblast growth factor receptor like 1,FGFRL1)等5个蛋白。FGFRs在成人体内具有重要的生理功能,包括器官发育、血管生成、细胞增殖和迁移、抗凋亡等。多项研究表明,FGFR的异常(包括基因的融合、重排、易位和扩增)会产生FGFR持续的自身二聚化和磷酸化,从而导致持续过度的FGFR激活,通过成纤维细胞生长因子受体底物2(fibroblast growth factor receptor substrate 2,FRS2)以及磷酸酯酶C-γ(phospholipase C-gamma,PLC-γ)激活下游丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)和磷脂酰肌醇-3激酶(phosphoinositide 3-kinase,PI3K)/蛋白激酶B(protein kinase B,PKB)信号通路。其他信号通路,如信号传导及转录激活因子(signal transducers and activators of transcription,STAT)也同样会被激活,过度激活的相关下游信号造成肿瘤发生发展相关基因的异常表达,其与癌症的发病和进展密切相关[9-10]。
其中FGFR2异常,尤其是基因融合突变(如BICC1,PPHLN1,CCDC6,MGEA5,TACC3),约占iCCA患者的20%,而在eCCA患者中几乎不出现[6,9,11-12]。大量临床前以及临床研究证明,靶向FGFR为CCA的治疗提供了一种新的、令人振奋的治疗策略[9]。FGFR抑制剂在有关CCA治疗方面的研究进展见表1。

表1 FGFR抑制剂有关胆管癌治疗的研究进展[13]Table 1 Research progress of FGFR inhibitors in cholangiocarcinoma treatment [13]

续表1
1.1.1 培米替尼培米替尼是一种强效选择性的口服FGFR1 ~ FGFR3抑制剂,为目前全球首款也是唯一一个获美国FDA批准的CCA靶向药物。该药由美国知名的全球性生物制药公司美国因塞特基因组公司开发,2020年4月被批准用于治疗成人晚期CCA(尤其是既往接受过治疗、含有FGFR2融合或重排、不可手术切除的局部晚期或转移性成人患者)。
在关键性的多中心Ⅱ期临床试验FIGHT-202 (NCT02924376)中[14],培米替尼(疗程为28 d:d1 — d21服药13.5 mg · d-1;d22 — d28停药)在107例有FGFR2融合或重排的患者中,二线治疗客观 缓 解 率(objective response rate,ORR)达 到35.5%,疾病控制率(disease control rate,DCR)高达82.2%,中位无进展生存期(median progressionfree survival,mPFS)为6.9个月,mOS达21.1个月;而临床一线标准治疗(吉西他滨+顺铂联合用药,CisGem)的mOS仅为11.7个月[15]。与靶点相关的高磷酸盐血症是最常见的1/2级不良事件(60%),通过控制磷酸盐摄入、使用磷酸盐结合剂以及利尿剂,可以控制不低于3级的不良事件中最常见的低磷血症(12%)。其他常见不良事件有脱发(49%)、腹泻(47%)、疲劳(42%)、指甲毒性(43%)和消化不良(40%)。另外,培米替尼可引起视网膜色素上皮脱离(retinal pigment epithelial detachment,RPED),说明书建议服药前及服药过程中进行全面的眼科检查。同时培米替尼可引起胎儿伤害,育龄患者需谨慎。
除了二线治疗外,培米替尼Ⅲ期临床研究FIGHT-302(NCT03656536)将头对头与一线标准治疗方案CisGem比较,评估培米替尼一线治疗有FGFR2基因融合或重排、不可手术切除的晚期或转移性CCA患者的疗效。
1.1.2 InfigratinibInfigratinib(BGJ398)是一种选择性口服FGFR1 ~ FGFR3抑制剂,由QED Therapeutics 公司从瑞士诺华公司获得其开发权。Ⅱ期临床试验(NCT02150967)数据显示,infigratinib(疗程为28 d:d1 — d21服药125 mg · d-1;d22 — d28停药)在一线治疗失败的FGFR2融合或其他变异的晚期或转移性CCA患者中,对FGFR2融合患者的ORR为18.8%,DCR为83.3%,对所有患者的预估mPFS为5.8个月,药效持续时间有限。试验期间25例患者(41%)报道了3级或4级治疗相关不良事件,其中包括高磷酸盐血症(16.4%)、口腔炎(6.6%)和手足综合征(4.9%)[16]。
目前,Ⅲ期PROOF临床试验(NCT03773302)正在进行,此项实验将在约350例有FGFR2基因融合或重排、不可手术切除的晚期或转移性成年CCA患者中进行,评估infigratinib与现有一线标准治疗方案CisGem对比的一线治疗疗效和安全性。2020年1月,FDA授予infigratinib一线治疗该类患者的快速通道资格认定。
值得注意的是,Krook等[17]以及Goyal等[18]研究发现,临床CCA患者使用infigratinib后出现FGFR2获得性耐药突变,包括FGFR2 N549K/H、V564F、E565A、L617V/M、K641R、K659M和K714R。临床前体外实验体系中也发现更多的类似获得性耐药突变类型,如FGFR2 M537I、V564I、K659E/N等,其他FGFR抑制剂(如Debio 1347)对这些突变不敏感[17,19]。
1.1.3 FutibatinibFutibatinib(TAS-120)是由大鹏药品工业和大冢制药共同研发的选择性FGFR1 ~ FGFR4不可逆抑制剂,其在体外对FGFR2野生型和多个获得性FGFR2突变(如FGFR2 N549K、L617V、K659M、N549H、E565A)敏 感。其 中N549K、L617V、K659M可导致infigratinib和Debio 1347活性降低12 ~ 39倍,而TAS-120仍对其保持较高的抑制活性[18]。2018年美国癌症研究学会(American Association for Cancer Research,AACR)大 会 摘要显示[20],infigratinibⅡ期临床试验中响应而后进展的3例FGFR2融合的iCCA患者中,2例在TAS-120的Ⅰ期临床试验(NCT02052778)中出现疾病缓解(partial response,PR),1例病情稳定(stable disease,SD)7.3个月[20]。BGJ398进展过程中的部分耐药突变在使用TAS-120治疗响应后恢复正常。2019年该临床试验再次报道了4例曾接受infigratinib或Debio 1347耐药的FGFR2融合的iCCA患者,服用TAS-120后,2例达到PR,2例SD 达5.1 ~ 17.2个月[18]。Ⅱ期临床FOENIX-101实验(NCT02052778)正在进行中。
除此之外,还有一些FGFR选择性抑制剂也在进行FGFR变异相关的CCA临床实验,如derazantinib(ARQ 087)、Debio1347、E7090等。
1.2 神经营养因子受体酪氨酸激酶抑制剂
NTRK由高度同源性的酪氨酸激酶受体A(tyrosine kinase receptor A,TRK-A)、TRK-B、TRK-C组成,由NTRK基因编码。NTRK基因若与其他基因融合,可导致TRK蛋白处于持续活跃状态,引发永久性的信号级联反应(包括下游PLC-γ、MAPK以及PI3K等信号通路的过度激活),导致细胞增殖、存活和侵袭异常,驱动TRK融合肿瘤的扩散和生长[21]。
NTRK基因是美国综合癌症网络(national comprehensive cancer network,NCCN)指 南 新 增的检测基因之一,NTRK融合突变较为罕见,CCA中频率约0.25%[22]、iCCA中约3.5%[23]。拉罗替尼(larotrectinib,LOXO-101)和恩曲替尼(entrectinib,RXDX-101)作为第1代NTRK抑制剂在NTRK融合阳性的患者中显示出快速而持续的临床响应,虽然患者中CCA患者例数稀少,但获得良好的初步结果[22,24]。美国临床肿瘤学会(American Society of Clinical Oncology,ASCO)2020年大会摘要显示,拉罗替尼在正在进行的针对携带TRK融合的成人肿瘤(包括CCA)患者的Ⅱ期篮式实验(NCT02122913、NCT02576431和NCT02637687)中,显示出惊人的药效,ORR高达71%,mPFS大于25.8个月[25]。同期,恩曲替尼也在进行类似的Ⅱ期试验(NCT02568267)。与FGFR抑制剂类似,第1代NTRK抑制剂临床应用中发现了基因突变导致的耐药现象,如TRK-A出现G595R突变、TRK-B出现G639R突变或者TRK-C出现G623R突变等。针对其耐药位点的第2代NTRK靶向药物LOXO-195目前正处于临床研究阶段,以期对抗第1代NTRK抑制剂用药后产生的耐药突变。NTRK抑制剂在CCA适应证上的研究进展见表2。
1.3 ALK 与ROS1抑制剂
ALK为间变性淋巴瘤激酶,其基因融合突变是非小细胞肺癌常见的一种驱动基因。而染色体重排也会导致ROS1与多种不同结合体发生融合,产生持续的激酶活性,导致细胞持续增殖引发肿瘤的发生,在CCA中的发生率约为8.7%[26]。ALK和ROS1激酶区域序列具有49%的同源性,在腺苷三磷酸(adenosine lriphosphate,ATP)结合位点则有高达77%的同源性,而存在的大多数差异发生在保守区域,因此推测ALK抑制剂可以用于ROS1变异的患者。ALK/ROS1抑制剂色瑞替尼(ceritinib)和克唑替尼(crizotinib)在CCA患者中正在进行相关的Ⅱ期临床试验(NCT02374489,NCT02034981)。另外,上文提到的NTRK抑制剂恩曲替尼同时具有ROS1/ALK抑制活性,目前也在ROS1/ALK融合的患者中进行相关药效和安全性的评估(NCT02568267)。

表2 NTRK抑制剂在胆管癌适应证上的研究进展[13]Table 2 Research progress of NTRK inhibitor in cholangiocarcinoma indications [13]
1.4 其他受体酪氨酸激酶抑制剂
除以上提及的靶点之外,对2007—2014年接受过人表皮生长因子受体2(human epidermal growth factor receptor 2,HER2/neu)靶向治疗(曲妥珠单抗、拉帕替尼或帕妥珠单抗),含HER2/neu基因异常或蛋白过表达的晚期CCA及GBC回顾性分析,9例GBC患者中3例SD,4例PR,1例CR。而CCA患者中HER2/neu突变比例更高,未发现影像学响应。因此在HER2阳性的CCA患者中单独或联合化疗使用靶向HER2的小分子药物仍需要进一步的临床数据支持[27]。其他RTK抑制剂在CCA适应证上的研究进展见表3。

表3 其他RTK 抑制剂在胆管癌适应证上的研究进展[13]Table 3 Research progress of other RTK inhibitor in cholangiocarcinoma indications [13]

续表3
2 异柠檬酸脱氢酶抑制剂
IDH1/2是柠檬酸循环中催化异柠檬酸转化为α-酮戊二酸(α-ketoglutaric acid,α-KG)的关键酶。IDH基因突变使催化合成α-KG的活性下降,却同时提高了促癌代谢物2-羟基戊二酸(2-hydroxyglutaric acid,2-HG)的生成,引发组蛋白以及DNA甲基化异常,从而导致肿瘤的发生发展[28]。
IDH抑制剂通过降低2-HG的异常产生而发挥作用,减少恶性细胞分化[29]。约有14%的iCCA患者携带IDH基因异常,而在eCCA患者中较少发生,且IDH1基因异常比IDH2更为常见[30]。
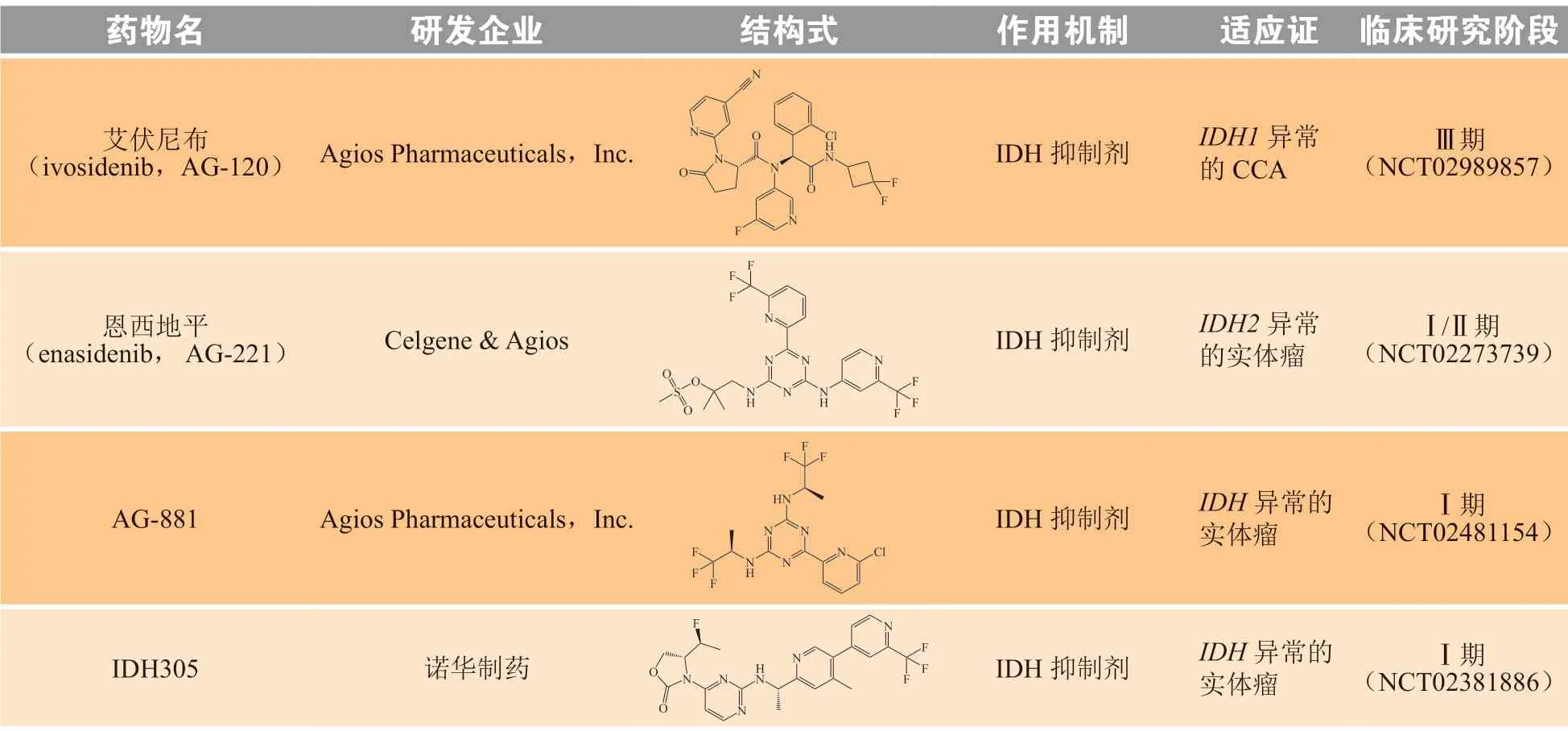
表4 IDH 抑制剂在胆管癌适应证上的研究进展[13]Table 4 Research progress of IDH inhibitor in cholangiocarcinoma indications [13]
2.1 艾伏尼布
艾伏尼布是一种针对IDH1突变的强效口服靶向抑制剂,由Agios Pharmaceuticals公司研发,被批准用于不能接受强化化疗的新诊断以及复发或难治性IDH1突变型急性髓细胞白血病(acute myelogenous leukemia,AML)患者。在既往化疗失败的73例IDH1突变晚期CCA的Ⅰ期研究(NCT02073994)中,结果显示艾伏尼布组的mPFS为3.8个月,mOS为13.8个月[31]。
关键性Ⅲ期ClarIDHy临床试验(NCT02989857)中,在185例接受过至少二线治疗并携带IDH1基因突变晚期胆管癌患者中,500 mg艾伏尼布(共计124例患者)mPFS为2.7个月(安慰剂组1.4个月),mOS为10.8个月(安慰剂组校正数据后为6.0个月)。艾伏尼布组ORR为2%,其中包括3例PR和63例SD;安慰剂组患者均未达到客观缓解,17例患者为SD[32]。艾伏尼布组常见的不良反应是腹水、恶心、乏力、腹泻等,整体耐受性较佳。艾伏尼布整体药效相较于FGFR抑制剂而言略逊色,但考虑到IDH和FGFR2变异的不重叠性,IDH抑制剂的开发对于含有IDH变异的患者亚群,仍具有不可或缺的临床意义。
2.2 恩西地平
恩西地平是一种针对IDH2突变的强效口服靶向抑制剂,由Celgene与Agios Pharmaceuticals共同研发,于2017年获美国FDA批准上市(商品名:Idhifa®),用于治疗含有IDH2突变的复发性或难治性AML患者,是首个针对肿瘤代谢的抗癌药物。其开展的Ⅰ/Ⅱ期临床研究,主要在有IDH2突变的实体瘤患者(包括iCCA患者,NCT02273739)中评估其有效性,虽然该试验已于2016年完成,但至今仍未有有关具体疗效的相关结果披露。
其他仍有一些IDH1、IDH2或泛IDH抑制剂也在不同的临床阶段(如NCT02481154和NCT02381886等)。关于IDH靶向抑制剂的获得性耐药机制,现暂不明确。研究推测,IDH不同亚型之间的互相代偿转化是可能的机制之一,如IDH1变异转向IDH2异常或反之亦然,IDH1获得性耐药的患者可能在使用IDH2抑制剂中获益[33]。
3 BRAF激酶抑制剂
BRAF是一种属于RAF蛋白家族的丝氨酸/苏氨酸蛋白激酶,该蛋白通过MAPK通路参与信号转导,刺激细胞的生长和存活。BRAF基因突变(V600E)可导致激酶活化,引发持续的信号通路激活,从而促进肿瘤发生。BRAF突变在CCA中主要发生在iCCA,占比约1% ~ 3%,80%以上为BRAF V600E突变[34]。目前已有针对BRAFV600E的靶向抑制剂,如维罗非尼(vemurafenib)、达拉非尼(dabrafenib)等。维罗非尼的一项Ⅱ期篮式实验(NCT 01524978)中,8例iCCA患者仅有1例患者部分缓解[35],推测其药效受限机制可能与其在结直肠癌中类似,与EGFR的反馈激活有关,联合MEK抑制剂可能有所改善[36]。达拉非尼联合使用曲美替尼(MEK抑制剂)在iCCA患者中显示持续的临床响应[37],在33例(32例可评估)CCA患者中(NCT02034110)ORR为41%,54%的患者缓解持续时间不小于6个月,mPFS为7.2个月,mOS为11.3个月[38]。

表5 BRAF抑制剂在胆管癌适应证上的研究进展[13]Table 5 Research progress of BRAF inhibitor in cholangiocarcinoma indications [13]
4 其他
除以上提及的,仍有一些CCA相关靶点处于早期研究中。对含有乳腺癌易感基因(breast cancer susceptibility gene,BRCA)突变的CCA患者临床实验进行回顾性分析,使用聚ADP核糖聚合酶(poly ADP-ribose polymerase,PARP)抑制剂的4例患者中,其中1例mPFS可达到42.6个月[39]。其他PARP抑制剂,如尼拉帕利和奥拉帕里布目前正在对含有异常DNA修复基因突变的CCA患者中进行疗效评估(NCT04042831、NCT03207347)。
Janus激酶 (Janus kinase,JAK)/STAT的异常激活在约50%的CCA患者中可以被观察到,STAT3的过度激活在iCCA患者中更为常见[40]。SphK-2抑制 剂Opaganib(Yeliva®,ABC294640),可以抑制STAT3的磷酸化激活,在Ⅰ期临床试验(NCT01488513)中初步显示出对CCA的阳性疗效。Opagani单用治疗晚期CCA的单臂Ⅱa期研究(NCT03377179)已达到预先设定的疗效目标(即12例患者中至少有1例响应),目前正在进行Opagani单用或联用硫酸羟基氯喹的入组及疗效评估。
在iCCA和eCCA患者中均发现了cMET的扩增和过表达,且其与生存周期的缩短有关[41]。但cMET抑制剂单药的临床疗效不佳[42],与化疗药物(吉西他滨)联用似乎更有希望[43]。
5 结语
吉西他滨联用顺铂依旧是临床CCA的一线标准治疗方案,该疗法的患者mPFS小于1年。越来越多的证据表明,针对CCA,特别是iCCA方向,临床涌现了更多的小分子靶向疗法的相关研究,如靶向FGFR2、IDH1/2、NTRK等。2020年,培米替尼获批成为全球首个明确用于治疗CCA的药物,突破了CCA多年“零靶向药物”的困境。NTRK也成为NCCN指南新增的检测基因之一,让具有该靶向基因突变的患者有药可用。总览中国CCA临床实验进展(Ⅰ/Ⅱ期),目前大多数为FGFR抑制剂,包括和记中国医药科技有限公司的HMPL-453、广东众生药业股份有限公司的ZSP1241以及培米替尼和E7090等。然而也需要关注随着靶向药物的临床使用而带来的耐药性问题。相信随着篮式实验的推广以及测序手段的发展,不久的将来,会有更多的靶向药获批用于CCA患者,大幅提高患者的生存获益。
猜你喜欢
传染病信息(2022年2期)2022-07-15
肝博士(2022年3期)2022-06-30
现代临床医学(2021年5期)2021-11-02
海外星云(2021年9期)2021-10-14
昆明医科大学学报(2021年4期)2021-07-23
现代仪器与医疗(2021年1期)2021-06-09
大众健康(2020年7期)2020-08-25
爱你(2019年13期)2019-11-14
科学24小时(2018年1期)2018-01-10
现代养生·下半月(2016年6期)2016-10-21
