
红外光谱法用于安钠咖中咖啡因和苯甲酸钠快速定性和定量分析
2021-03-30 09:43:54苏承轲刘翠梅孟鑫花镇东段凯
法医学杂志 2021年1期
苏承轲,刘翠梅,孟鑫,花镇东,段凯
1.鄂尔多斯市公安局,内蒙古 鄂尔多斯017000;2.公安部禁毒情报技术中心 毒品监测管控与禁毒关键技术公安部重点实验室,北京100193
现有的安钠咖中咖啡因的定性方法主要为显色法[1]和气相色谱-质谱法,定量分析方法主要为滴定法[1]和高效液相色谱法[4]。定性方法中显色法准确度低,气相色谱-质谱法检测速度慢、成本高。定量方法中滴定法操作繁琐,高效液相色谱法操作繁琐、检测速度慢、成本高。基于衰减全反射(attenuated total reflectance,ATR)的红外光谱法基本无需样品前处理,测试速度快、检测成本低、绿色环保,已被用于毒品、易制毒化学品、新精神活性物质的快速定性和定量检测[5-8]。本研究在分析大量缴获安钠咖样品红外光谱图的基础上,挑选咖啡因和苯甲酸钠的特征吸收峰,建立采用红外光谱法定性分析安钠咖中咖啡因和苯甲酸钠的方法;同时,采用混合制样的方法制备定量建模样本,建立采用红外光谱和偏最小二乘法(partial least squares,PLS)定量分析安钠咖中咖啡因和苯甲酸钠的方法。
1 材料与方法
1.1 主要仪器与试剂
FrontierTMFT-IR/NIR 光谱仪配备金刚石光窗的单次衰减全反射附件(美国PerkinElmer 公司),LC-20A 高效液相色谱仪(日本岛津公司),Agilent Zorbax Eclipse XDB-C18色谱柱(4.6 mm×150 mm,5 μm;美国Agilent 公司)。Scientz-15DZ 高通量组织研磨器(宁波新芝生物科技股份有限公司),HW450AS 型远红外干燥箱(北京科伟永兴仪器有限公司),XS 105 电子天平(美国梅特勒-托利多公司),KQ 3200E 超声波清洗器(昆山市超声仪器有限公司),Lab Dancer涡旋振荡器(德国IKA公司)。直径3mm金属球用于研磨使用。
甲醇、乙腈(色谱纯,德国Merk公司)、三乙胺(色谱纯,国药集团化学试剂有限公司)、浓磷酸(含量≥85%,色谱纯,国药集团化学试剂有限公司)。乙醇(分析纯,国药集团化学试剂有限公司)。咖啡因[阿拉丁试剂(上海)有限公司,纯度99%],苯甲酸钠(国药集团化学试剂有限公司,纯度99%)。48 份缴获安钠咖样品由鄂尔多斯市公安局提供。
1.2 样品制备
将缴获的安钠咖样品于70 ℃烘箱干燥2 h,记录烘干前和烘干后质量。取适量样品和1个金属球装入5 mL 的塑料离心管中,采用高通量组织研磨器对其进行研磨混匀,研磨功率为70 Hz,研磨时间为2 min。
将高纯度咖啡因和苯甲酸钠按不同质量比例混合,配制成咖啡因纯度(质量分数)分别为7.5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、95%的17 个样品。配制样品的质量约为0.2 g。将称量好的样品和一个金属球装入5 mL 的塑料离心管中,采用高通量组织研磨器对其进行研磨混匀,研磨功率为70 Hz,研磨时间为2 min。
1.3 傅里叶变换红外光谱
取研磨均匀的固体样品适量,均匀地铺展在ATR窗口的上表面,压紧使紧密接触,波数范围4 000 cm-1~550 cm-1,分辨率4 cm-1,采样次数16 次。采集时间小于1 min。
红外光谱图的采集和定量建模分析采用Spectrum 10.5.3和Spectrum Quant软件10.5.3(美国PerkinElmer公司)。
果蔬运输系统作为服务行业,应用物联网技术对提升整体管理水平,使果蔬运输服务在速度上和质量上得以体现,使得运输过程中数据的传输更加准确及时,便于交互、监控并及时反馈信息。
采用SPSS 17.0.1 软件(美国IBM 公司)进行配对样本t检验分析。
1.4 PLS定量模型
采用全波段光谱进行计算,选取标准正态变量(standard normal variate,SNV)校正联合一阶导数的光谱预处理方法,建立PLS 定量分析模型,计算模型的决定系数(R2)、交叉验证均方差(root mean square error of cross validation,RMSECV)、预测均方差(root mean square error of prediction,RMSEP)。
计算测试样品与所建PLS 定量模型的马氏距离值,马氏距离值小于3 时表明测试样品与建模样品的基质极为相似,定量结果可靠[9]。
选取一个咖啡因和苯甲酸钠混合样品(咖啡因纯度为35%)进行PLS 定量模型的重复性和重现性考察。将该样品在1 d 内连续测定6 次,计算6 次测定结果的相对标准偏差(relative standard deviation,RSD),作为重复性数据;将6 份样品分别在不同的6 d 中进行测定,计算6 次测定结果的RSD,作为重现性数据。
1.5 高效液相色谱分析
Agilent Zorbax Eclipse XDB-C18色谱柱(4.6 mm×150 mm,5 μm),柱温35 ℃,流速1.0 mL/min,进样量5 μL,检测波长240 nm,流动相A 相为乙腈,流动相B相为磷酸-三乙胺缓冲液(浓磷酸4.12 mL 被200 mL 超纯水稀释,取三乙胺5.56 mL 滴入磷酸溶液中,再加入水稀释至1 000 mL,混匀后超声脱气备用)。梯度洗脱:0 min 15%A,0~5.0 min 15%~80%A,5.0~5.2 min 80%~90%A,5.2~7.0 min 90%A,7.0~7.2 min 90%~15%A,7.2~10.0 min 15%A。咖啡因的保留时间为3.02 min,苯甲酸钠的保留时间为4.64 min。
准确称取安钠咖样品5.00 mg 于15 mL 塑料离心管中,加入10 mL 甲醇,放入超声波清洗器中超声10 min,辅助溶解,以5 000×g离心10 min,取1 mL 进LC-20A 高效液相色谱仪分析。
准确称取咖啡因和苯甲酸钠标准品5.00 mg 于10 mL 玻璃容量瓶中,加入甲醇定容至10 mL,超声10 min 溶解,配制成0.5 mg/mL 的标准样品溶液,取1 mL 进LC-20A 高效液相色谱仪分析。
1.6 傅里叶变换红外光谱和高效液相色谱定量方法的比较
选取咖啡因纯度在25%~50%的9 个实际缴获安钠咖样品,分别采用高效液相色谱和傅里叶变换红外光谱进行定量分析,对两组数据进行配对样本t检验。检验水准α=0.05。
2 结果与讨论
2.1 傅里叶变换红外光谱结果
咖啡因的傅里叶变换红外光谱图(图1A)中1 693、1 644 cm-1为C=O 双键的伸缩振动,1 547 cm-1为C=N 双键的伸缩振动,1 481、1 454 cm-1为N-CH3的反对称变角振动,1 429、1 404 cm-1为N-CH3的对称变角振动,1 358、743 cm-1为环状酰亚胺结构的伸缩振动,1 285~1 024 cm-1为C-N 键的伸缩振动。
苯甲酸钠的傅里叶变换红外光谱图(图1B)中1 596 cm-1为苯环的伸缩振动,1 548、1 406 cm-1为C=O的伸缩振动,706 cm-1为苯环的弯曲振动。
图1C 为咖啡因与苯甲酸钠混合样品(咖啡因纯度50%)的傅里叶变换红外光谱图,图中咖啡因和苯甲酸钠的红外吸收峰明显可见。混合样品中咖啡因的1 693 cm-1和1 644 cm-12 个吸收峰的位置发生了较大的偏差,变为1 698 cm-1和1 650 cm-1,这是因为纯咖啡因样品中这2 个峰为宽峰,而混合样品中变为窄峰,峰形发生了变化,所以峰的位移也发生了较大变化。
图1D 是咖啡因和苯甲酸钠混合物(咖啡因纯度10%~80%)的红外光谱图。咖啡因的6个吸收峰1 698、1 650、1 237、972、743、609 cm-1和苯甲酸钠的6 个吸收峰1 596、1 548、1 406、845、708、679 cm-1在所有的混合物样品中均检出,因此将上述峰分别定为咖啡因和苯甲酸钠的特征吸收峰。
图1E 是48 份安钠咖缴获样品(未经干燥处理)的傅里叶变换红外光谱图。分析结果表明,所有样品中均检出了咖啡因(1 698±8)、(1 650±8)、(1 237±3)、(972±4)、(743±3)、(609±3)cm-16 个吸收峰和苯甲酸钠(1596±3)、(1548±3)、(1406±3)、(845±3)、(708±3)、(679±3)cm-16 个吸收峰。
多数缴获安钠咖样品性状潮湿,从图1E 中也可以看出,超过三分之二的安钠咖缴获样品在900~600 cm-1出现基线下移,这是典型的含水样品的特点(图1F)。但这些潮湿样品的基线下滑并不影响其特征峰的检出。
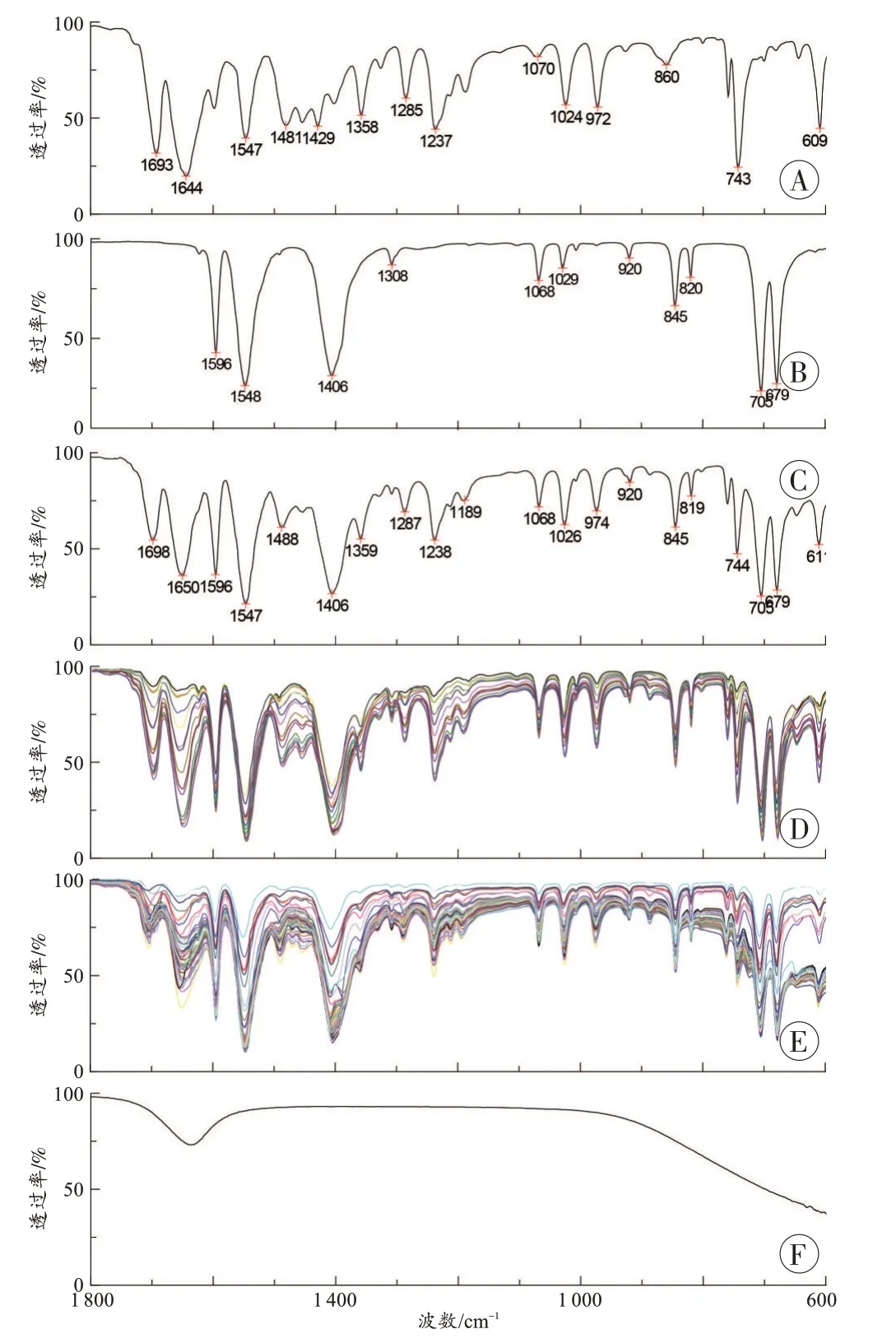
图1 ATR-傅里叶变换红外光谱图Fig. 1 ATR-Fourier transform infrared spectra
2.2 特征吸收峰法的定性检出限
图2 为咖啡因纯度分别为7.5%和10%的2 个混合样的红外光谱图,当咖啡因的纯度为7.5%时,检出了咖啡因的5 个特征吸收峰和苯甲酸钠的6 个特征吸收峰,咖啡因的1 650 cm-1峰未检出。当咖啡因的纯度为10%时,咖啡因和苯甲酸钠的6 个特征吸收峰均检出。在苯甲酸钠纯度为5%的混合样品中检出了苯甲酸钠的6 个特征吸收峰。因此,采用特征吸收峰为定性判别依据时,安钠咖中咖啡因和苯甲酸钠定性分析的检出限分别为10%和5%。
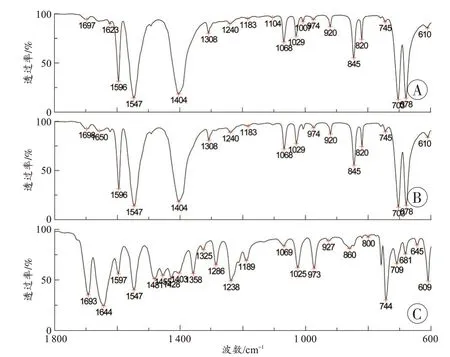
图2 咖啡因与苯甲酸钠混合物的ATR-傅里叶变换红外光谱图Fig. 2 ATR-Fourier transform infrared spectra for mixture of caffeine and sodium benzoate
2.3 PLS定量分析模型的建立
采用全波段光谱进行计算,选取SNV 联合一阶导数的光谱预处理方法,建立PLS 定量分析模型。所得到的标准曲线见图3。咖啡因PLS 定量模型的线性范围为10%~80%,R2为99.9%、RMSECV 为0.68%,RMSEP 为0.91%;苯甲酸钠PLS 模型的线性范围为20%~90%,R2为99.9%、RMSECV 为0.91%、RMSEP 为1.11%。
为了保证定量结果的准确性,定量建模样品和实际样品的性状不能有太大的差异。由于配制的定量建模样品是干燥的,而多数缴获样品性状潮湿,所以,实际缴获样品在采集光谱前需要先进行烘干处理。烘干时记录烘干前后质量的变化。
经测定,PLS 定量分析方法中咖啡因的重复性为0.39%、重现性为1.28%,苯甲酸钠的重复性为0.67%、重现性为2.40%,可见所建定量模型的测定精度高。
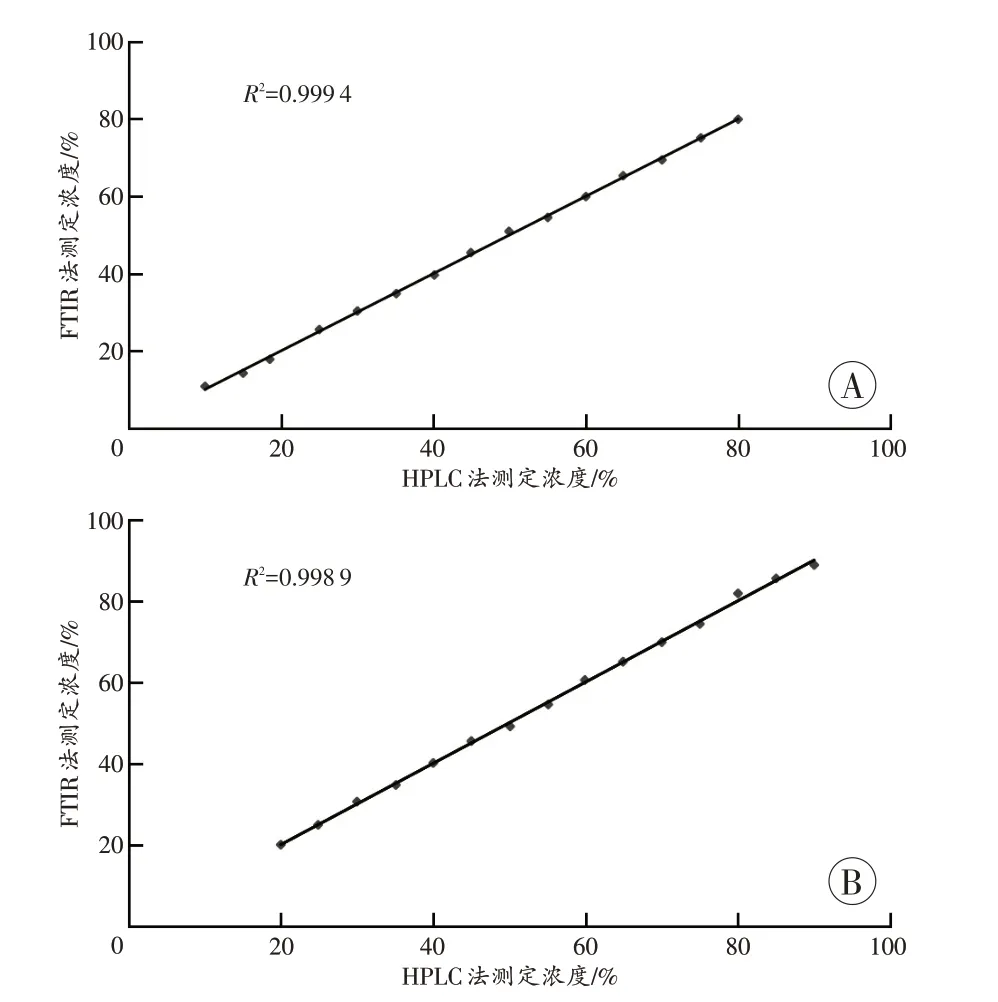
图3 PLS定量模型标准曲线Fig. 3 PLS quantitative model standard curve
2.4 傅里叶变换红外光谱和高效液相色谱定量方法的比较结果
选取咖啡因纯度在25%~50%的9 个实际缴获样品,分别采用高效液相色谱和傅里叶变换红外光谱进行定量分析,结果见表1。对2 组数据进行配对样本t检验,傅里叶变换红外光谱和高效液相色谱测定的咖啡因纯度平均值分别为39.6%和38.1%,差异无统计学意义(P>0.05);傅里叶变换红外光谱和高效液相色谱测定的苯甲酸钠纯度平均值分别为60.4%和63.0%,差异无统计学意义(P>0.05)。因此,傅里叶变换红外光谱的测定结果准确可靠。
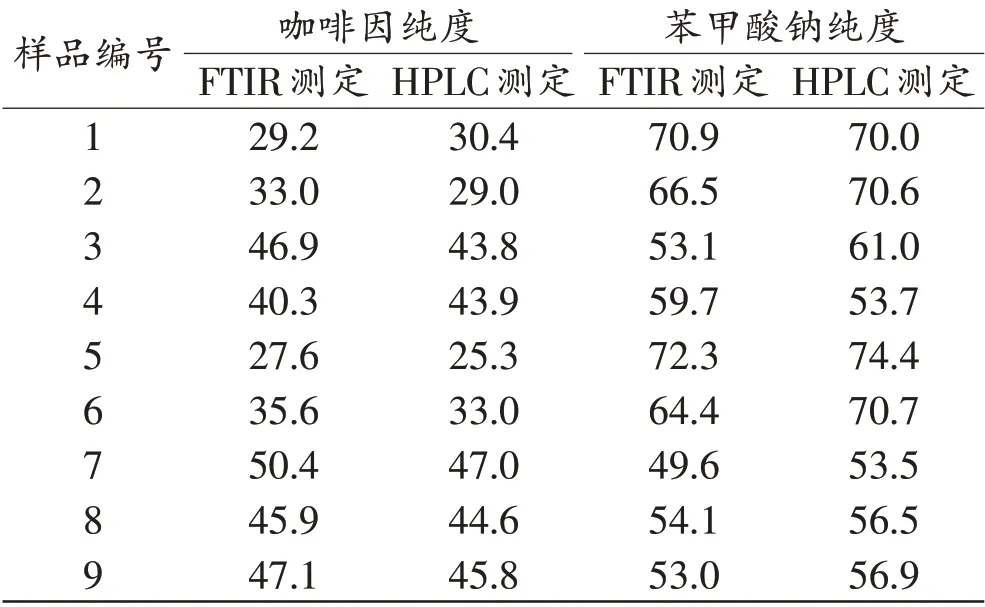
表1 安钠咖样品中咖啡因和苯甲酸钠傅里叶变换红外光谱和高效液相色谱测定纯度结果比较Tab. 1 Comparison of the determined purity of caffeine and sodium benzoate in Annaca samples by Fourier transform infrared spectrum and high performance liquid chromatography (%)
2.5 安钠咖缴获样品测定
采集48 份缴获样品的傅里叶变换红外光谱数据,采用特征吸收峰法对样品进行定性分析,所有样品均检出了咖啡因和苯甲酸钠。为了保证待测样品与建模样品的基质一致,在进行定量分析前需要对样品进行干燥处理,干燥后样品与建模样品中咖啡因之间的马氏距离值0.03~1.30,干燥后样品与建模样品中苯甲酸钠之间的马氏距离值0.02~1.20。干燥后样品计算得到的马氏距离值均小于3,说明测试样品与建模样品的基质极为相似,因此计算所得定量结果可靠。48 份干燥样品中咖啡因的纯度为27.6%~63.1%,平均值为41%;苯甲酸钠的纯度为36.9%~72.3%,平均值为59.0%。需要说明的是,某些实际缴获样品中可能会掺入其他物质,导致计算得到的马氏距离值大于3,此时采用高效液相色谱定量方法进行定量分析结果会更加准确。
2.6 结论
本研究建立了安钠咖样品中咖啡因和苯甲酸钠快速定性和定量分析的傅里叶变换红外光谱方法,确定了咖啡因和苯甲酸钠的6 个特征吸收峰,以全部特征吸收峰均检出为阳性判断依据时,咖啡因的定性检出限为10%。建立了基于红外光谱的PLS 定量模型,模型线性好,检测精度高。采用配对样本t检验法对高效液相色谱和傅里叶变换红外光谱的测定结果进行了比较,结果表明,两个方法的定量结果之间差异无统计学意义。采用所建立的傅里叶变换红外光谱定量方法分析了48 份安钠咖缴获样品,样品中咖啡因的纯度为27.6%~63.1%、苯甲酸钠的纯度为36.9%~72.3%。
猜你喜欢
腐蚀与防护(2023年10期)2023-11-28 09:59:16
表面技术(2023年2期)2023-03-06 02:44:02
数学物理学报(2019年2期)2019-05-10 11:32:38
测控技术(2018年7期)2018-12-09 08:58:26
大自然探索(2017年10期)2017-10-28 06:47:59
大自然探索(2017年5期)2017-05-26 17:48:07
实用临床医学(2016年8期)2016-06-07 01:28:16
舰船科学技术(2016年1期)2016-02-27 15:39:21
电测与仪表(2015年5期)2015-04-09 11:30:44
郑州大学学报(医学版)(2015年1期)2015-02-27 14:50:35
