
应用HID Ion GeneStudioTM S5 测序系统探讨毛干样本线粒体DNA异质性
2021-03-30 09:43程凤张庆霞陈成建李万婷张家榕张更谦严江伟
法医学杂志 2021年1期
程凤,张庆霞,陈成建,李万婷,张家榕,张更谦,严江伟
1.山西医科大学法医学院,山西 太原030001;2.北京市公安司法鉴定中心,北京100192
线粒体DNA(mitochondrial DNA,mtDNA)与核DNA 相比,由于较高的拷贝数量,检测样本的需求量低,对降解的现场生物检材,如毛发、陈旧尸骨、指甲等样本的检测具有优势[1]。但目前绝大多数情况下仅对mtDNA 高变区Ⅰ和mtDNA 高变区Ⅱ进行双脱氧链终止法(dideoxy chain-termination method)检测,由于所检测突变位点的数量有限,使其难以准确将无关个体进行区分[2]。随着测序技术的不断发展,以规模化平行测序为主的二代测序技术逐渐应用于法庭科学领域[3]。
由于二代测序技术具有可对mtDNA 进行多样本多位点同时检测的优势[4],使mtDNA 个体识别能力得到一定的提高[5]。本研究使用Precision ID mtDNA Whole Genome Panel试剂盒(美国Thermo Fisher Scientific 公司)[6]对线粒体全基因组进行扩增,应用HID Ion GeneStudioTMS5 测序系统(美国Thermo Fisher Scientific 公司)对线粒体全基因组分析检测,并对其分型结果进行探讨,为法庭科学的应用提供参考。
1 材料与方法
1.1 样本收集
按照“知情同意”原则,分别采集8 名汉族无关个体的口腔拭子,静脉血及额、顶、颞部、会阴部毛发样本各1 份,每份毛发样本包含2 根毛发。样本总共为48 份。
1.2 样本处理
将口腔拭子和静脉血涂卡后室温放置,晾干后常温储存。对毛发样本使用以下步骤进行处理[7]。首先,剪取距离毛根2 cm 处长度为2 cm 毛干样本,使用200 μL 1%十二烷基硫酸钠(sodium dodecyl sulfate,SDS)溶液浸泡,以离心半径10 cm,1 000 r/min,离心5 min,弃去上清液。加入200 μL 纯水浸泡,以离心半径10 cm,1 000 r/min,离心5 min,弃去上清液。再使用400 μL 无水乙醇浸泡10 min 后弃去所有上清液,晾干后常温储存。在实验操作过程中,为避免交叉污染,需将不同个体的检材样本分批次处理。
1.3 DNA提取
使用QIAamp DNA Investigator 试剂盒(美国QIANEN 公司)[8],遵照实验步骤分别提取口腔拭子、血卡和毛干全基因组DNA。为了评估样本中所含mtDNA的情况并保证后续文库的成功构建,本研究使用自行设计的扩增片段分别为364 bp 的线粒体特异性引物(正向:5′-CACCTACACCCCTTATCCCC-3′;反向:5′-CATTAGGAGGGCTGAGAGGG-3′)和682 bp 的线粒体特异性引物(正向:5′-TCGGAGGACAACCAGTA AGC-3′;反向:5′-GCACTCTTGTGCGGGATATT-3′)对所有样本进行扩增检测。
1.4 文库构建
采用Precision ID mtDNA Whole Genome Panel(美国Thermo Fisher Scientific 公司)对线粒体全基因组进行目的片段DNA 扩增,将已知线粒体基因组分型DNA 样本作为阳性对照,灭菌去离子水作为阴性对照。为减少测序后所产生冗余序列的影响,应用FuPa 试剂(美国Thermo Fisher Scientific 公司)对多余引物进行消化。分别采用Ion TorrentTMPrecision ID Library 试剂盒(美国Thermo Fisher Scientific公司)和Applied BiosystemsTMPrecision ID IonCodeTMBarcode Adapters 1-96 Kit in 96-Well PCR Plate(美国Thermo Fisher Scientific 公司)进行标签序列和测序接头的连接。最后使用AgencourtTMAMPure XP磁珠(美国Beckman Coulter 公司)和70%乙醇溶液进行纯化。
1.5 文库定量
对构建完成的线粒体全基因组文库使用7500 Real-Time PCR 系统(美国Thermo Fisher Scientific公司)和Ion TorrentTMIon Library TaqManTMQuantitation 试剂盒(美国Thermo Fisher Scientific 公司)进行定量。
1.6 测序和数据分析
将定量后的文库稀释至40 pm/μL,使用Ion TorrentTMHID Ion ChefTM系统(美国Thermo Fisher Scientific公司)进行测序模板制备。将制备的测序模板使用Ion S5TMPrecision ID Chef & Sequencing 试剂盒(美国Thermo Fisher Scientific 公司)在HID Ion GeneStudioTMS5 测序系统进行测序,每次初始化运行2 次。共使用3 张芯片Ion 530TMChip(美国Thermo Fisher Scientific 公司)。使用快速质量控制软件FastQC-0.11.7(Simon Andrews 开发)[9]对数据进行质量控制。以修正剑桥序列为参考序列,使用ConvergeTM软件v2.1(美国Thermo Fisher Scientific 公司)对测序数据初步分析。此外,使用SAM tools 1.0[10]对测序结果中的BAM(binary map format)文件进行详细分析,同时使用Morpheus 在线软件(https://software.broadinstitute.org/morpheus)对不同样本的线粒体全基因组不同区段的测序深度进行对比。根据ZHOU 等[11]的报道,将次要等位基因频率定为大于15%,测序覆盖度大于100×作为本次实验异质性位点的筛选条件,同时使用整合基因组学查看器IGV-2.3(James T. Robinson 开发)[12]进行测序结果的可视化展示,并对异质性位点进行双脱氧链终止法验证。
2 结 果
使用扩增片段分别为364 bp 和682 bp 的线粒体特异性引物对48 份样本进行扩增检测,均检测出特异性扩增产物。48 份样本测序文库的定量范围为3.26~5.32 ng/μL,均符合测序检测要求。3 张芯片产生原始测序数据总量1.44 G,平均有效测序量达40%,其中文库平均连接率为98.6%,多克隆文库为39.5%,低质量文库为15.5%,有效文库为52.3%,共11 011 759 个读取片段,片段平均读长为129 bp。口腔拭子的平均测序深度为(1 970±690)×,血卡的平均测序深度为(2 062±1 118)×,毛干样本平均测序深度为(1 159±743)×(各个样本的详细测序深度见附表1)并且阳性对照结果正确,阴性对照中未发现污染存在。
使用ConvergeTM软件v2.1 将测序数据与参考序列进行比对分析,共获得119 个变异位点,其中6 个位点位于高变区Ⅰ,15 个位点位于高变区Ⅱ,98 个位点位于其他区域,各个样本变异位点的详细分型见附表2。8 名个体的变异位点数目分别为29、40、38、35、13、36、40 和35。48 份样本测序覆盖度为(102~3 898)×,其中毛干样本的测序覆盖度较低,每个样本的测序覆盖度见图1。通过查看样本所有位点的测序覆盖度情况,发现在所有样本中nt8674~nt8762 区段的测序覆盖度均小于500×。当次要等位基因的筛选频率大于15%时,在个体1 中颞部毛干样本的nt9756 位点(T/C)和个体3 中颞部毛干样本的nt16354 位点(T/C)出现异质性,并与双脱氧链终止法验证结果一致(图2A~B),其余6 名无关个体的口腔拭子、血液及不同部位毛干样本的线粒体全基因组分型结果相同。当次要等位基因的筛选频率降至10%时,在多名个体的nt16390、nt9540、nt8020 等位点出现异质性。然而对其进行双脱氧链终止法验证,结果均为假阳性。例如,在个体8 颞部毛干样本的nt16390 位点次要等位基因A 频率为13%,双脱氧链终止法验证结果中只有G 的基因分型(图2C)。
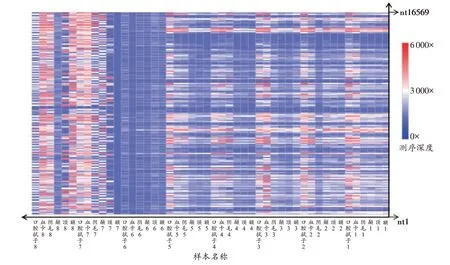
图1 样本的测序覆盖度Fig. 1 Sequencing coverage of samples

图2 3名个体颞部毛干样本的二代测序IGV图(上)与双脱氧链终止法结果图(下)Fig. 2 Second-generation sequencing IGV maps of three individual temporal hair shaft samples(upper)and the diagrams of the dideoxy chain-termination method(lower)
3 讨 论
mtDNA 多态性的研究主要集中在非编码区和部分编码区,其中位于非编码区的高变区Ⅰ(nt57~nt372)和高变区Ⅱ(nt16024~nt16383),目前为法医学线粒体检测最主要的区域。张幼芳[13]对100 名浙江汉族无关个体的高变区Ⅰ进行分析,仅发现71 种单倍型,84 个变异位点,平均每个个体仅出现0.84 个碱基变异。张明阳等[14]通过对150 名随机个体的高变区Ⅱ进行分析,仅发现9 个单倍型。这意味着高变区mtDNA遗传多态性有限,无法用于个体同一性认定,而使用二代测序技术对线粒体全基因组进行检测,可以同时对多位点样本进行检测,从而获得更多的mtDNA 信息。本研究使用Precision ID mtDNA Whole Genome Panel(主要采用叠瓦式的扩增方法对全线粒体基因组进行扩增,包含平均扩增片段长度为163bp的162对引物)对8 名无关个体进行全线粒体基因组测序,共发现119 个变异位点,额外获得除高变区以外的98 个碱基变异信息。
KING 等[15]使用二代测序技术对线粒体全基因组进行检测后认为,当位点的测序覆盖度大于40×时,其分型结果具有参考价值。本研究使用线粒体特异性引物对所有样本进行扩增检测,均检测出特异性扩增产物,提示成功提取到线粒体基因组DNA,并且满足全线粒体基因组短片段扩增的需求,可用于后续文库的构建和测序。在本研究中,所有样本的平均测序深度均大于100×,证明此次测序结果较为可靠。相对于口腔拭子和血卡,毛干样本属于微量降解样本,构建文库浓度较低,在进行文库定量或纯化等操作步骤时导致部分文库的损失,使其测序覆盖度相对其他种类样本较低。同种类样本之间的测序覆盖度标准差大于500×,可能是由于不同个体检材样本之间的扩增效率差异,从而造成样本间平均测序覆盖度差异较大。由于在文库的构建过程中,可能会因为扩增引物结合位点发生突变或者由于扩增区域中含有较多的单碱基重复[16-18]等原因,导致扩增效率不均一,从而形成各个测序位点覆盖度之间的差异,例如,在本研究所有样本中nt8674~nt8762 区段,线粒体测序覆盖度均相对较低。本研究将次要等位基因频率定为大于15%时,筛选出2 个异质性位点,并与双脱氧链终止法检测结果一致。但当将次要等位基因频率降至10%时,若干位点的二代测序结果均为异质性结果,却未能通过双脱氧链终止法检测进行验证。出现这种结果的原因可能为:(1)由于二代测序中扩增错误或测序错误等原因所产生的碱基变异;(2)相对于二代测序技术,双脱氧链终止法检测的灵敏度较低,未能检测出低比例的碱基变异。
mtDNA 的异质性是影响法医线粒体基因分析的重要因素之一,在案件检测中应引起重视。在同一个体中存在2 种及以上的线粒体基因序列,称为mtDNA的异质性,主要分为2 种情况:(1)同一组织含有2 种及以上的线粒体基因序列;(2)同一个体不同组织中存在2 种及以上的线粒体基因序列。目前对于异质性的出现及其机理尚不明确,但对与线粒体基因相关疾病的研究具有重大意义[19]。使用二代测序技术对线粒体全基因组进行检测,在增加其检测范围及测序覆盖度的同时,也增加其异质性检出的可能。二代测序技术的灵敏度较高[20],导致由于污染、扩增错误以及测序错误带来的碱基差异更容易被检测,另外复合扩增体系中的简并引物设计以及短片段扩增所引起同源部分的核DNA 片段的扩增,也会对检测结果产生一定的影响[21]。因此在使用二代测序技术对法医mtDNA 进行分析时,应谨慎对待出现的异质性,为避免假阳性对检测结果的影响,需注意以下几点:(1)严格实验操作,避免污染;(2)采用高保真聚合酶,降低扩增错误;(3)应尽量增加测序覆盖度,减少简并引物对结果产生干扰;(4)假如出现疑似异质性分型位点,使用双脱氧链终止法检测进一步验证;(5)应根据样本的测序情况设置相应的次要等位基因频率分析阈值,避免阈值过低而出现较多假阳性结果。
猜你喜欢
临床肺科杂志(2022年3期)2022-11-26
科学技术创新(2022年30期)2022-10-21
中华实用诊断与治疗杂志(2022年1期)2022-08-31
科学技术创新(2021年35期)2022-01-14
中国卒中杂志(2021年7期)2021-11-29
绿色科技(2021年6期)2021-04-17
孔学堂(2020年1期)2020-06-01
西安科技大学学报(社会科学版)(2020年1期)2020-04-01
中华诗词(2019年1期)2019-11-14
中国体育科技(2018年6期)2018-12-13
