
聚偏氟乙烯多铁性的第一性原理研究
2021-03-29 02:42陈亦杰胡俊杰胡立峰胡盼盼
湖北第二师范学院学报 2021年2期
杨 辉,陈亦杰,胡俊杰,胡立峰,胡盼盼
(湖北第二师范学院 a.物理与机电工程学院;b.湖北省环境净化材料工程技术研究中心,武汉 430205)
1 引言
多铁性材料是一种具有多种铁性特征的磁电耦合多功能材料,它可以直接通过电场调控磁性能,具有超低能耗的突出优势以及灵活的电场调节能力。这在开发新型多功能电子器件方面展现出巨大潜力,如磁电存储器,多铁性隧道结,高灵敏度磁场探测器,可调电感,以及高频可调微波器件[1]-[3]等等。
有机多铁材料的研究近几年来获得了一系列的研究成果。Thoms等人[4]使用 Pariser-Parr-Pople模型建模,通过数值研究方法,发现当甘菊环低聚物数在8个以上时,结构会表现出铁磁性基态,随后的电荷密度计算表明电偶极子取向导致低聚物具有铁电性。杨辉等人[5]结合第一性原理和量子蒙特卡罗方法,研究了不同碳位置氢化的三五富瓦烯和五七富瓦烯低聚物,结果表明氢化后的低聚物具有室温铁磁性。Herrero等人[6]在石墨烯表面沉积单个氢原子,使石墨烯可以在较长距离上维持铁磁性,表明实验上可以通过 STM 对氢原子进行原子精度的操控,来控制石墨烯区域的磁性。显然,有机多铁材料以其丰富物种,性能多样和无毒性重金属稀土元素等特点成为多铁材料研究的重要方向。最近,Ma.Y.W.等人[7]通过裁剪的方法,在 Teflon表面引入碳原子悬挂键,发现裁剪表面处碳悬挂键之间产生自旋交互作用,Teflon表现出室温铁磁性,这引起了我们的关注。
与Teflon结构非常相近的聚偏氟乙烯(PVDF)是非中心对称结构,具有电极化特性,其电极化强度为153μC/m2。能否通过裁剪方法,在聚偏氟乙烯分子链上诱导磁性能,从而设计出具有室温多铁性的聚偏氟乙烯材料。本文采用裁剪方法在聚偏氟乙烯表面引入碳原子悬挂键,通过第一性原理方法,计算聚偏氟乙烯分子链的磁性质和电极化性质,从理论计算的角度给出了材料的基态磁性质,着重分析其磁性产生原因和磁电耦合强度。
2 计算方法
本文选用计算软件包Materials Studio中的castep模块,包含基于密度泛函理论(DFT)[8]的平面波赝势(PAW)方法,并采用Perdew-Burke-Ernzerhof进行广义梯度近似(GGA)[9](用于离子电位的PBE)用于模拟电子-电子和电子-离子相互作用。采用能量截止值为450 eV的平面波基组和3×3×1 Monkhorst-Pack 点网格进行几何结构优化。更精细的5×5×1k点采样方案用于自洽计算。能量和最大力的收敛标准分别设定为10-5eV和0.01eV/Å。每次计算都采用2×1×1的超级单元。
计算中使用β相PVDF晶体参数通过实验给出。结晶PVDF具有由线性链组成的伪六方结构。在我们的第一原理计算中,首先关注PVDF剪切后,含有C-H悬挂键的PVDF分子链。 PVDF分子链总共有10层,沿z轴的真空厚度为15Å,足以避免相邻异质结构之间的相互作用,结构如图1所示,其中黑色表示碳原子,白色表示氢原子,蓝色为氟原子。我们的计算产生的平衡C-C键长为1.56Å,键合能为3.46 eV,这与报道的值是一致的[10]。
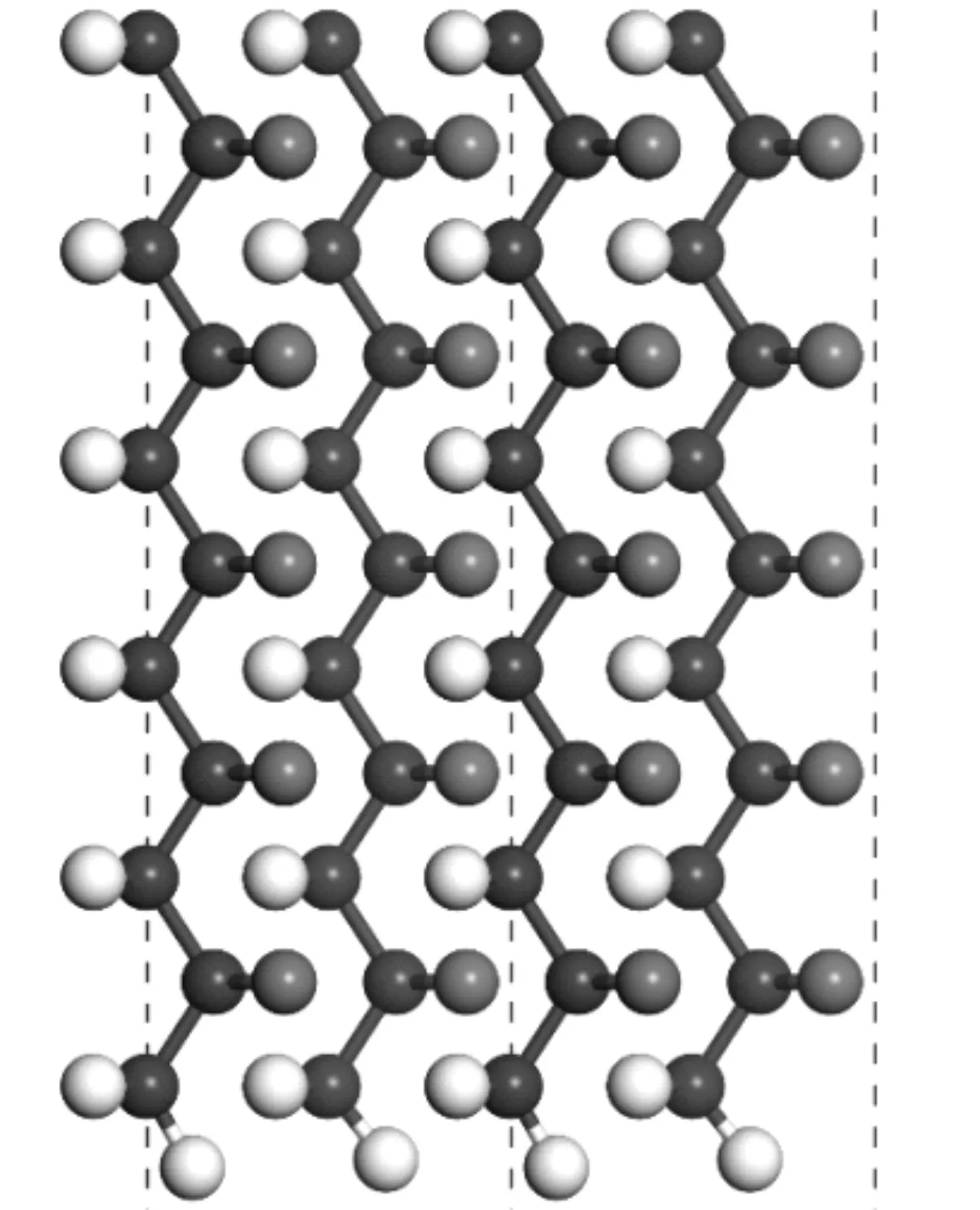
图1 聚偏氟乙烯裁剪后晶体结构图
3 结果讨论和分析
3.1 磁性研究
由于结构存在未饱和键,不同交换关联势对计算的结果可能存在较大影响。在研究结构性能之前,我们首先测试不同交换关联势参数对计算结果的影响。选取实验PVDF结构链间距为4.59Å,我们分别计算了四种不同的交换关联势GGA,LDA,VDW和HSE在铁磁基态下以及反铁磁基态下结构的能量。为了探讨结构剪切后是否具有磁性,通过计算结构的铁磁与反铁磁的能量来进行比较,如果铁磁态的能量小于反铁磁态的能量,此时认为该剪切结构具有铁磁性质。计算结果如表1。

表1 不同交换关联势下结构的能量(meV)
表1的结果显示出PBE,VDW和HSE这三种不同的交换关联势的计算结果之间的差异相对较小。其中,最大差值在0.31 meV范围,但LDA的计算结果与PBE,VDW和HSE的结果差异较大,最大差值为1.98 meV,表明PBE,VDE和HSE这三种交换关联势的计算均具有一定的准确性。我们在以后的所有计算中都使用PBE方法。 另一方面,从铁磁态与反铁磁态的能量差(EFM-EAFM)的计算结果可以看出,其值为负数,表明当具有碳悬挂件的PVDF链间距在平衡位置4.59Å时,结构具有磁性质,磁性基态为铁磁态,而Teflon在平衡位置时,并不具有磁性质,这与Teflon的计算结果不同。
考虑到在实际剪切方法中,切面会产生一定的应力,影响表面结构。为了更好地描述真实剪切情况下PVDF结构磁性能,我们计算截面应力对结构磁性能的影响。以实验PVDF结构链间距4.59Å为基准,以表面应力对结构可能存在拉伸或压缩为依据,应变变化幅度以0.2%为一个应变单位,计算不同的应变变化下,结构磁性质与链间距的关系。计算结果如图2所示。
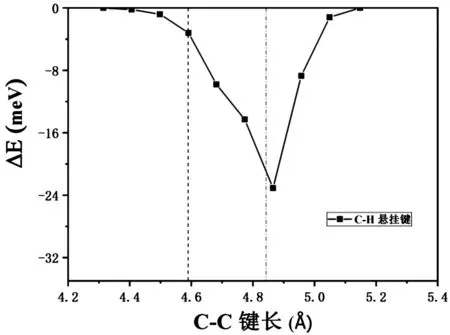
图2 能量差(铁磁态与反铁磁态)与PVDF链间距的关系
在图2中,两条虚线分别为实验测量PVDF链间距4.59Å和计算结构弛豫的最佳链间距4.84Å,实线为应力影响下,能量差(铁磁态与反铁磁态)与PVDF链间距的关系。根据ΔE的计算结果,可以看出,当PVDF链间距在4.4Å和5.08Å之间时,ΔE<0,表明此时铁磁态能量低于反铁磁状态,铁磁态是最低能量磁性基态,结构表现出铁磁性。当链间距约为4.86Å时,铁磁态和反铁磁状态能量ΔE差达到-25 meV。当在250K时,-ΔE大于25meV热力学激励能量标度时,室温下的热波动不能破坏铁磁基态。因此,在当-ΔE>25 meV的含有C-H悬挂件PVDF链中可以预期近室温铁磁性。
为了进一步探究含有C-H悬挂键PVDF链的电子结构,我们通过GGA方法计算获得了具C-H悬挂键结构PVDF链的态密度,如图3所示。首先,我们可以清楚地看到C-H悬挂键PVDF链有带隙,带隙约为1.9 eV,表明该结构是半导体。在费米表面附近,上下自旋状态出现明显的劈裂,表明该结构处于铁磁状态,这与之前ΔE的计算结果一致。为了更好地描述结构磁交换性质,我们计算了结构的自旋密度,自旋密度图如图4所示,其中深色表示电子自旋向上,浅色表示电子自旋向下。从图4中,我们可以看到碳悬挂键之间的铁磁耦合。自旋密度主要集中于剪切的PVDF链面上的碳原子,并且它们之间通过氟原子相互耦合。表面碳原子自旋密度沿PVDF链延伸到碳原子中,但迅速衰减。 同时,这些状态在能量上很好地局域化。
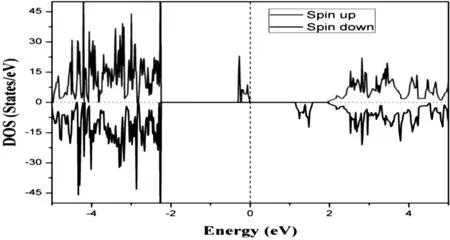
图3 具有C-H悬挂键结构PVDF链态密度图
这些结果表明,即使PVDF分子链发生应变,由于铁磁耦合主要位于相邻PVDF链中的C悬挂键之间,并且很难改变相邻PVDF链中C悬挂键的构型以形成共价键,因此PVDF中的铁磁态相对稳定。

图4 PVDF分子链自旋密度图
3.2 电极化性能研究
为了探究PVDF材料的电极化特性,基于最大局域化瓦尼尔函数的理论,首先计算各原子的波恩有效电荷。Wannie[9]函数能够准确地表明结构中各原子轨道以及原子间杂化轨道的信息(如s、p、d轨道等),是研究处理电子空间局域的有效方法。Wannier函数定义为倒空间中固体能带结构Bloch波函数的傅里叶变换
(1-1)
式子(1-1)中,WnR(r)表示实空间中第n个价带的Wannier函数,φmk表示为倒空间中第m个价带的Bloch波函数,积分范围是第一布里渊区。这里,各离子的波恩有效电荷是通过使用wannier函数来求解。
进一步,根据自发极化公式,计算结构自发极化强度:
(1-2)
其中(ni)和ri代表第i个碳位点上的电荷及其坐标。 表示与每个碳位点的平均电荷的偏差。计算结果如表

表2 PVDF晶体和具有C-H悬挂键PVDF 分子链自发极化强度结果
由表2的结果可以看出, PVDF晶体的电极化强度为153μC/m2,这说明其本身具有电极化性质。进行剪切处理之后,C-H悬挂键结构的电极化强度为196μC/m2,可以得知剪切之后PVDF的电极化性能发生了改变。剪切之后增强了晶体结构的电极化强度,使得PVDF晶体结构同时具有磁性和铁电性,表明该结构具有多铁性。另一方面,从表2中可以看出PVDF的电极化强度变化幅度在43μC/m2左右,变化幅度非常小,同时磁性能较弱,表明通过裁剪方法引入磁性时,材料电极化性能略微增强,结构磁电耦合性能非常弱。
4 结论
通过第一性原理计算方法研究了PVDF分子链的磁性和铁电性能。结果表明,PVDF晶体处于非磁性状态,而磁性状态可以通过裁剪产生具有碳悬挂键的结构引出磁性质,预测C-H悬挂键的PVDF中存在室温铁磁性。磁性起因于截面处碳自旋局域化,自旋密度沿PVDF链延伸向碳原子中,但迅速衰减。PVDF分子链磁性的引入同时增强结构的电极化性能,结构存在磁电耦合。我们的研究不仅表明,具有C-H悬挂键的PVDF是非常具有潜力能达到室温有机多铁性的材料,也为有机多铁材料的设计提供了新思路。
猜你喜欢
数学物理学报(2022年5期)2022-10-09
军民两用技术与产品(2020年2期)2020-03-14
发明与创新·小学生(2019年11期)2019-08-11
军事文摘·科学少年(2017年4期)2017-06-20
小天使·三年级语数英综合(2017年6期)2017-06-07
小天使·三年级语数英综合(2017年6期)2017-06-07
学生天地·小学低年级版(2016年9期)2016-05-14
中学科技(2015年6期)2015-08-08
火炸药学报(2014年3期)2014-03-20
船海工程(2013年5期)2013-01-11
