
1例成骨不全患者致病基因鉴定
2021-03-22 00:30黄泳华冯穗华冼诗瑶潘焯仪吴欣新张妙莲吴海涛广东省江门市中心医院广东江门529000
广东医科大学学报 2021年1期
赵 强,黄泳华,冯穗华,冼诗瑶,潘焯仪,吴欣新,张妙莲,吴海涛(广东省江门市中心医院,广东江门 529000)
成骨不全(OI)是一种以骨量降低、骨骼脆性增加、反复骨折及蓝巩膜为主要特征的单基因遗传病,在新生儿中发生率约为1/20000[1-2]。成骨不全临床表型异质性和遗传异质性都非常强,临床表型谱非常丰富,不同患者临床表现差异非常大,可以仅表现为骨密度稍低,也可有严重骨骼异常,甚至出现婴儿期致死[3-4]。1979年Sillence根据临床表现和遗传方式将其分为 4种亚型[5],随着基因检测技术的发展,成骨不全相关致病基因被陆续鉴定发现[6],目前按致病基因分类已达十余种。通过检索OMIM及 Pubmed数据库可发现,目前已报道与成骨不全临床表现明确相关基因已有 10余个,其中大多数已报道病例均是由常染色体显性基因COL1A1和COL1A2所引起[7],COL1A1和COL1A2为Ⅰ型胶原蛋白编码基因。近年来不断有报道由常染色体隐性基因突变所导致的成骨不全病例[8],这些基因可调节Ⅰ型胶原蛋白的转录后修饰、分泌及加工[2]。由于各亚型在临床表现及影像学特征上的重叠,成骨不全的临床诊断及分类一直都比较困难[9]。随着近年高通量基因测序技术在临床诊断上广泛应用,尤其适应于成骨不全这类临床表现度及遗传异质性均较复杂的疾病分子诊断。在本研究中,我们对1 例疑似成骨不全的家系使用相关基因捕获测序方法,对测序数据进行生物信息学过滤、筛选及分析,鉴定该成骨不全家系致病性基因突变,并对基因突变进行家系遗传机制分析及相关功能学预测研究,以期为其临床诊断、预防及后续生育阻断提供有效的分子诊断信息。
1 病例资料
1.1 研究对象
患儿为 10岁女孩,自4 岁起反复骨折,查体及X线检查显示双下肢及前臂弯曲畸形,胸骨畸形,双下肢及肱骨多处骨折(图 1),无明显压痛,伴有牙发育不良,蓝色巩膜(图 2),听力正常。临床诊断为疑似成骨不全,但一直未进行相关基因检测。患儿父母均无类似临床表现,自述无家族史。本研究采集患儿及父母外周血,并采用本地 50名正常个体外周血作为对照。本研究获得医院伦理委员会审核批准,所有研究对象均征得本人或监护人同意,并签署知情同意书。
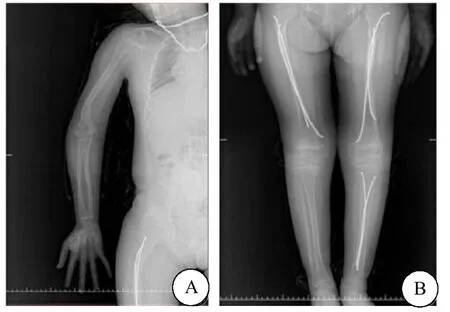
图1 患儿 X射线照
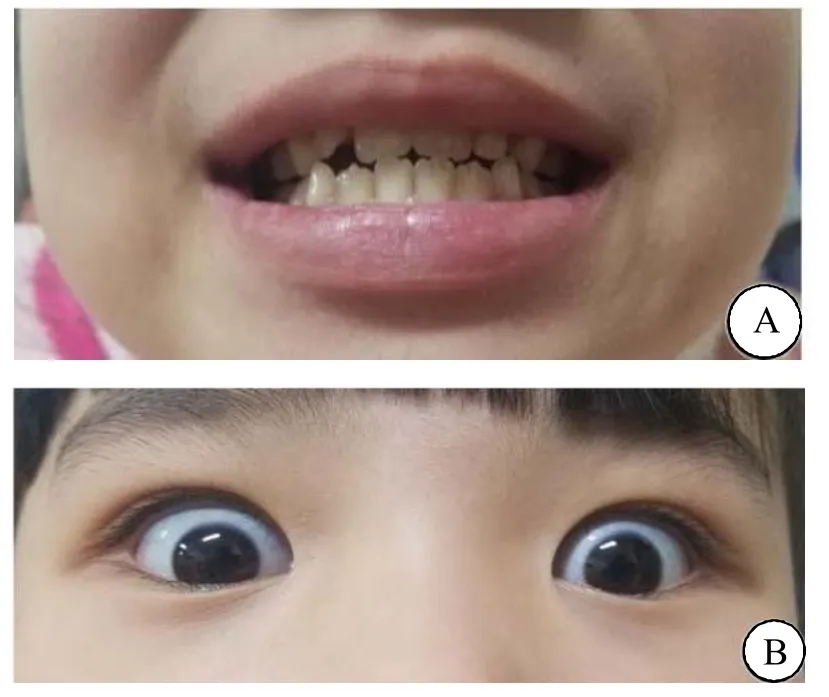
图2 患儿巩膜及牙齿发育临床表现
1.2 标本处理
使用5 mL EDTA抗凝管采集患儿及其父母外周血,使用DNA提取商品化试剂盒(QIAamp DNA Blood Midi Kit,Qiagen,Germany),按产品说明书提取各研究对象DNA。
1.3 成骨不全相关基因捕获测序
使用超声波仪将提取的基因组DNA打断为约100~500 bp小片段,然后通过磁珠筛选打断后DNA片段,筛选出主片段大小为150~200 bp,末端补平后在 3' 端加碱基“ A”采用华大基因(BGI,Shenzhen,China)定制芯片与患者DNA库于47℃杂交16~24 h,该芯片包含 13个成骨不全目标基因编码区及临近剪接位点区捕获探针(表 1),杂交结束后进行探针洗涤和洗脱反应。文库经片段大小、浓度检测,合格后各文库进行 pooling、定量,然后行单链环化。环化后的文库经过制备形成 DNA纳米球,利用高通量测序仪 BGISEQ-500(BGI,Shenzhen,China)连续双向测序。
1.4 测序下机数据分析
下机原始数据(Raw reads)进行测序质量评估,去除低质量以及被接头污染的reads[10]。随后用BWA软件(Burrows Wheeler Aligner) 与HG19人类基因组数据库进行序列比对[11],GATK软件进行SNV(single nucletide mutation) 和Indel(insertion and deletion)鉴定,生成目标区域碱基多态性结果,随后进行数据库(NCBI dbSNP,HapMap,1000 human genome dataset和database of 100 Chinese healthy adults)比对,并对鉴定出的可疑突变进行注释及筛选,突变致病性评估是基于ACMG流程[12]。
1.5 Sanger测序验证
使用Oligo7软件针对疑似致病突变及周边区域设计扩增引物,对患儿及其父母基因组DNA进行突变区域特异性 PCR扩增,扩增产物经纯化后,进行Sanger测序反应,判断二代测序鉴定出的疑似致病突变是否真实,且突变是否符合遗传规律。
1.6 基因突变生物功能分析和预测
通过国内外文献检索、致病突变数据库(HGMD和Clinvar)及突变后对基因表达翻译的影响判断突变功能影响。通过Human Splicing Finder、Maxant Scan及NNSplice软件预测内含子突变对 mRNA剪接的影响及产生新剪接位点可能性。
2 结果
2.1 基因捕获高通量测序及数据过滤
患儿经过 13个成骨不全致病基因捕获测序,获得了相关基因外显子区及邻近 15个碱基的内含子区域的碱基序列片段,经一系列生信过滤分析,测序数据各质量参数均合格见表1。
2.2 候选基因突变过滤筛选
鉴定出单碱基突变及 20个碱基以下缺失插入突变共 22个,经过分析筛选流程,最终筛选出P3H1基因中一对复合杂合突变为患儿疑似致病突变,分别为c.2164C>T(p.Q722*) 和c.466-7T>G,其中突变c.466-7T>G未有文献及数据库报道,属于新突变,见表2。
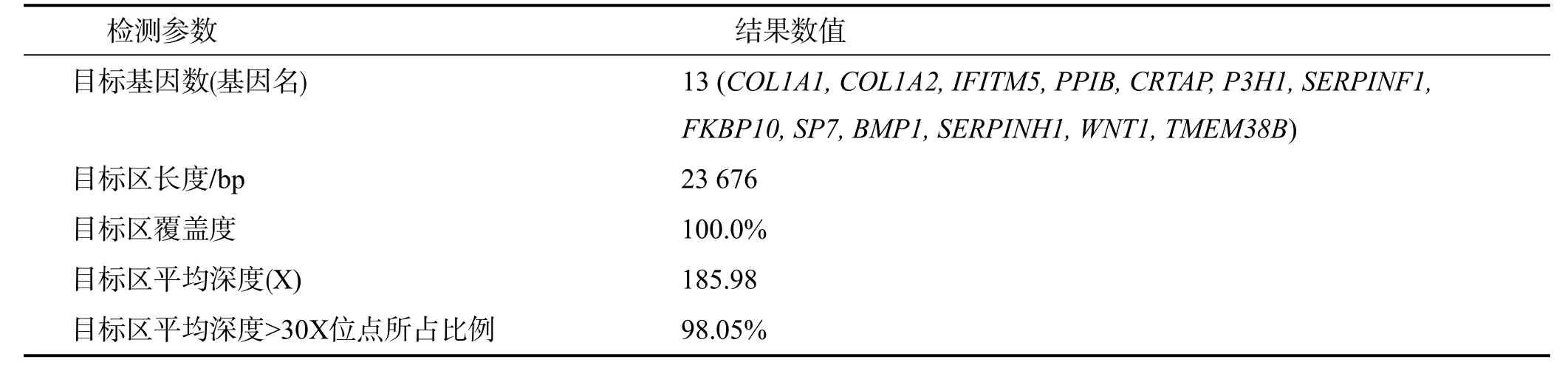
表1 测序数据质量参数值

表2 候选致病变异筛选流程
2.3 家系Sanger测序验证
针对c.2164C>T(p.Q722*)和c.466-7T>G位点设计PCR扩增引物,对患儿及其父母行 PCR扩增,扩增产物进行 Sanger测序验证,其中突变c.2164C>T(p.Q722*)来源母亲,突变 c.466-7T>G来源于父亲,见图3。
2.4 生物信息分析及预测
突变 c.2164C>T(p.Q722*)为无义突变,导致722位氨基酸提前产生终止密码子,使得722位及后续氨基酸无法翻译,P3H1蛋白发生截断。而突变c.466-7T>G位于内含子区域,经预测该突变导致新剪接位点产生及原剪接位点破坏可能性较大。其中Human Splice Finder软件评估野生型和突变后的CV (consensus value)分别为56.84和85.79(阳性阈值为 65),ΔCV为+50.93%(阳性阈值为+10%)。其中Maxant Scan软件评估野生型和突变CV(consensus value)分别为-2.32和6.26(阳性阈值为 3),ΔCV为+369.83%(阳性阈值为+30%),见图4。
3 讨论
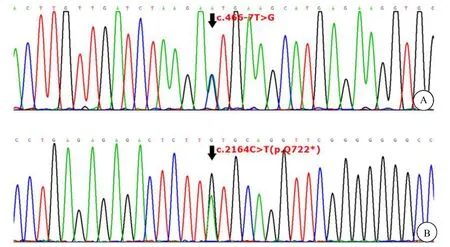
图3 家系中P3H1 c.466-7T>G 和c.2164C>T (p.Q722*) Sanger测序图
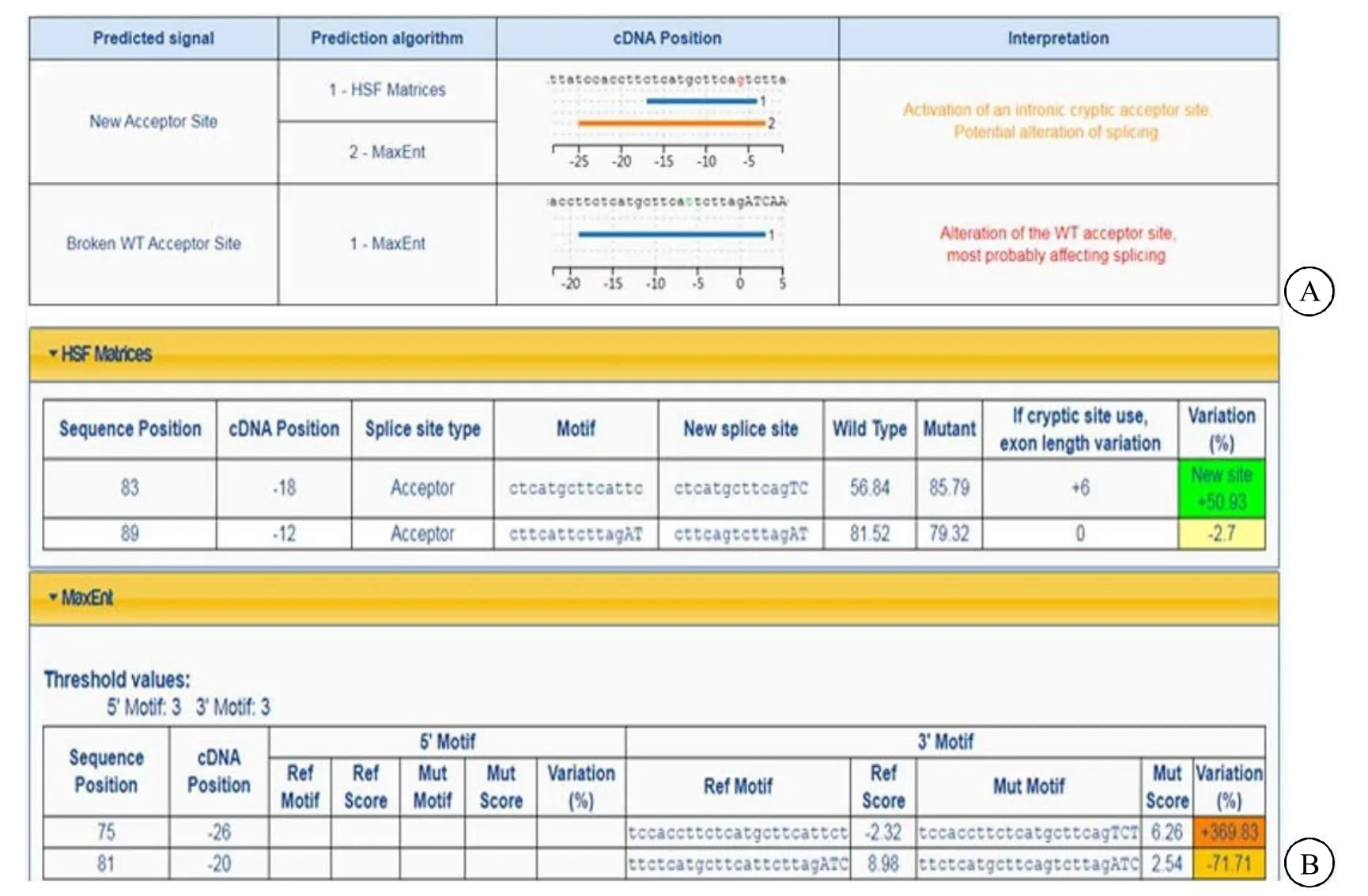
图4 突变c.466-7T>G基因剪接功能预测
P3H1基因编码脯氨酰-3-羟化酶-1,该蛋白属于参与胶原生物合成、折叠及组装的胶原羟化酶家族成员,它与CRTAP蛋白、PPIB蛋白组成胶原蛋白翻译后修饰复合体,负责对I型胶原蛋白单个脯氨酸残基进行羟基化[13-14]。P3H1是一个常染色体隐性致病基因,该基因纯合突变或复合杂合突变可导致成骨不全 VIII型,通过检索 HGMD及 Pubmed数据库,目前已报道的P3H1致病性突变已有 60多个,其中无义突变、错义突变及剪接位点突变占大多数。本研究对疑似成骨不全患者使用成骨不全相关基因捕获测序,通过生物信息分析及筛选,鉴定出 P3H1基因复合杂合突变 c.2164C>T(p.Q722*) 和c.466-7T>G为疑似致病性突变,患儿其余 12个成骨不全基因内未发现符合遗传机制的突变。
其中 c.2164C>T(p.Q722*)为无义突变,2017年Huang等[9]报道该突变 c.2164C>T(p.Q722*)和c.105_120del(p.D36Rfs*16) 为1 例产前成骨不全Ⅷ型胎儿的致病突变,他们通过Western Blot实验显示携带该突变患儿 P3H1蛋白表达缺失,而 mRNA表达水平并无显著降低。Cabral等[13]报道P3H1的无义突变会导致终止密码子提前出现,可能通过无义突变介导降解机制使得mRNA和蛋白表达水平降低。Chang等[15]也通过Western blots和免疫荧光显微镜检测技术,证实P3H1无义突变可导致蛋白表达降低或缺失,而mRNA表达水平是正常的。通过上述研究可判断突变c.2164C>T(p.Q722*)影响 P3H1蛋白表达,为成骨不全VIII型致病性突变。
另一突变 c.466-7T>G位于P3H1的 1号内含子剪切位点区域,通过检索 HGMD数据库及文献均未报道该突变,属于新突变。通过检索数据库及文献,发现多篇文献报道P3H1基因剪切位点区域突变为成骨不全 VⅢ致病性突变,其中大多数为剪切供体和受体突变,还有3个病例研究报道致病突变位于剪切位点深部区域(如:c.1170+5G>C、c.1473+3A>C和c.2055+18G>A)[16-18],其中 c.2055+18G>A突变可导致 mRNA表达降低及新的剪接体出现。本研究通过多个剪接位点预测软件对突变 c.466-7T>G分析,均提示该突变很可能导致新剪接方式产生,从而影响该基因转录和翻译。对该患儿进行随访,其父告知患儿已去世,后续未能继续进行体内功能学研究。
本研究对1 例疑似成骨不全患儿使用 13个成骨不全致病基因高通量测序,鉴定出P3H1基因内 2个复合杂合突变,在本地 50例正常对照(100条染色体)中均未发现这 2个突变。其中 c.2164C>T(p.Q722*)为已报道的致病性突变,而 c.466-7T>G未有文献报道和数据库收录,但有文献报道P3H1基因内含子内突变影响基因剪切,导致成骨不全 VⅢ型。本研究通过目前主流基因剪切位点突变功能预测软件,推测该突变影响P3H1基因的剪切,导致蛋白构造及功能发生改变。由于人体内功能学实验无法进行,后续条件允许情况下,可进一步在体外细胞和动物水平研究该突变相关功能。
猜你喜欢
口腔医学(2021年10期)2021-12-02
矿产勘查(2020年11期)2020-12-25
航空发动机(2020年3期)2020-07-24
中成药(2017年12期)2018-01-19
中华老年口腔医学杂志(2016年2期)2017-01-15
西安建筑科技大学学报(自然科学版)(2016年1期)2016-11-08
湖南畜牧兽医(2016年3期)2016-06-05
铁道科学与工程学报(2015年4期)2015-12-24
中国病理生理杂志(2015年8期)2015-12-21
天津护理(2015年4期)2015-11-10
